
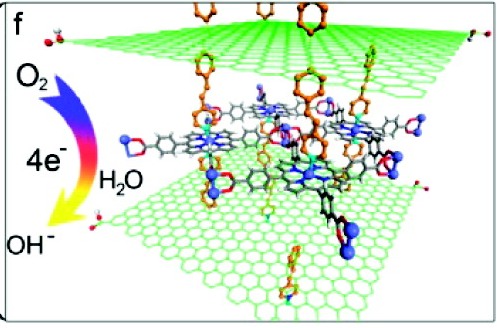

Title: Electrocatalytically Active Graphene–Porphyrin MOF Composite for Oxygen Reduction Reaction
DOI: 10.1021/ja211433h
Author: Maryam Jahan, Qiaoliang Bao, and Kian Ping Loh
Journal: Journal of the American Chemical Society
Affiliation: Graphene Research Centre, Department of Chemistry, National University of Singapore, Singapore
Take-home Importance According to the Authors: By reacting the pyridine-functionalized graphene with iron–porphyrin, a graphene–metalloporphyrin MOF with enhanced catalytic activity for oxygen reduction reactions (ORR) is synthesized. The structure and electrochemical property of the hybrid MOF are investigated. The results show that the addition of pyridine-functionalized graphene changes the crystallization process of iron–porphyrin in the MOF, increases its porosity, and enhances the electrochemical charge transfer rate of iron–porphyrin, showing a facile 4-electron ORR and can be used as a promising Pt-free cathode in alkaline Direct Methanol Fuel Cell.
Take-home Importance According to the Blogger: Composite material that takes advantages of each component, metalloporyphrin’s high reactivity with metal-organic framework’s high porosity. As someone works in adjacent fields, this concept is fairly inevitable. I am glad someone finally made it work and I applaud their effort (and I will be using something else for my third year original research proposal).
Tidbit from the Blogger: Sorry for the delay as this article is intended a month ago for April, but there is the Ph.D. qualification oral exam, a matter which I have to attend to. Either way, I intend a double-issue with a following post, so hopefully your hunger for science (or just the commentary) can be satisfied.
If you have not heard already, there is a recent (for me at least) movement for open access of journals, which is probably prompted by the mathematics community against the publishing giant Elsevier (one example here by Time Growers). Although the lack of subscription to the Journal of Electroanalytical Chemistry is to my dismay, I got over it after checking the library fact sheet. As many research is funded by the public by taxpayers, I am all for open access of those work or even a small fee to fund the management so the general public can clearly see where their support went to. However, for the chemistry community in particular, I believe open peer-review of research articles may be a separate but more immediate goal. When you became a graduate student and started to check literature for references (at least in my field in particular), you began to realize that not all published procedures are reproducible if followed word-by-word (such as adding 1 L of solvent to a 10 mL reaction vessel). Many review the peer-reviewing process as an irritating duty and mistakes were probably made on both sides. But having a larger body of scientists reviewing a pre-published work and commenting on the tricks in utilizing a post-published work can be extremely beneficial. Maybe this idea can be adapted eventually. Anyways, as always, thanks for listening.
Summary: There are several “hot” topics in material and inorganic chemistry nowadays: 1) catalytic small-molecule transformation on a metal center (complexes such as metalloporphyrins), 2) porous materials and their modifications as high-surface-area scaffolds (polymers such as metal-organic frameworks, or MOFs), 3) electron and redox process management bridged by highly conductive structures (conjugated carbon derivatives such as graphene). Combine everything together, an efficient electrocatalyst is born. Earlier works by Hupp and coworkers have established that metal-organic frameworks (or MOFs) with metalloporphyrins as building units can sustain energy-transfer interactions with other ligands in the same molecule. Equally as important, they have constructed a system where the active metal centers do not bind to the building units and are free to interact with small-molecule targets. Thus, it is expected for the community to explore more interesting structures that can enhance the catalytic process.

A very typical coordination scheme in MOFs is a di-metal-tetracarboxylate-paddlewheel unit where the axial position on the cluster nodes are coordinationally unsaturated if the metal species present are divalent. For example, Zn2(COOR)4 construction motif is typical, while higher oxidation states may involve metal-metal bonding or axial ligand binding to provide charge balance. In this case, by appending benzenecarboxylate groups (a common strategy in porphyrin-based MOF construction), individual porphyrins can be linked and fixed into a square grid via coordination chemistry. However, in this case, the so-called MOF is an aggregation of two-dimensional sheets of linked porphyrin, not truly a three-dimensional framework. However, as earlier works have shown, these two-dimensional sheets can be easily linked together by 4,4′-bipyridine neutral basic linkers that will coordinate to the unsaturated axial sites and self-assemble to form a three-dimensional structure. A pretty neat trick that is, and the authors here take advantage of that linkage motif by diazotizing the reduced-graphene oxide (rGO) sheets with 4-(4-aminostyryl) pyridine to provide the lock and key necessary for a three-dimensional structure.
All said and done, it is a quite interesting structure. The internal space of the rGO is now expanded by the introduction of porphyrin sheets (which I disagree with author’s claim that the graphene existed as a structural impurity, as additional powder x-ray diffraction peaks were observed. Observed x-ray diffraction peaks are definite indications of structural changes that may result from highly organized local structures, which then should not be considered as an impurity. Seriously, those formations are the heart of this study).
The expanded sheets should allow easier access to catalytically active prophyrins. Besides in-depth characterization with infrared spectroscopy (functional group mapping) and UV-vis absorption spectroscopy (electronic structure mapping, as well as doping concentration correlation), the more interesting properties–the electrochemical properites–are characterized in depth with cyclic voltammetry, with ferrocyanide control and then with oxygen reduction in 0.1 M KOH. There is indeed an increase in current density when greater amount of iron-porphyrin was doped in the system.


Great! Interesting structures and interesting properties.
Well, maybe.
Although the concept is great, there are a lot of experimental details that may sound more amazing than what they actually are (IN MY OPINION). Let’s start with the Support Information.
1) As I have mentioned before, considering the paddlewheel structure formed, divalent metal ions will charge balance with the carboxylate and allowing binding with neutral pyridine. If the metal is not divalent, then the basic pyridine groups will be less likely to bind comparing to a charged ion like chloride. As I sort through the information that was swpet under the rug, the starting material is ferric chloride [Fe(III) Cl3], which means the axial positions on the paddlewheel are likely occupied by chloride ions. If not, there there must be some counter ions hanging out, somewhere. Now if you check the synthetic scheme above, hmmm… of course porphyrin will be a metal-scavenger and take up excess iron in solution. However, if the paddlewheel positions are occupied, then the most Lewis acid sites would be directly on the porphyrin iron, which deactivate the catalytic activity of the porphyrins.
And then, your catalytic cycle became a little bit messy.
Also, the structure analysis by XPS alone was insufficient (considering the raw data was not very smooth to start, further analysis is stretchy to say at the least). Of course two iron environments are expected within the structure with difference in electron density, but that is about all XPS can show. There is no information on how iron coordinate in this composite material. To convince a chemist that the pyridine actually coordinates to iron, the shift in binding energy is not enough (considering strong pi interaction between the porphyrin sheets, which presumably is nonexistent in the composite). The oxidation state problem needs to be addressed if not with Mossbaur spectroscopy or EPR. Then, if you really want something more definite, write a proposal for EXAFS.
2) Potentiostat never reads current density directly, which means a scientist has to manually input the surface area of the electrode. See the absorption isomer plateau that correlates with surface area, then see the current density. As much as I trust the authors corrected for the increase in surface area due to expanded graphene surface from MOF intercalation, it seems a bit too close. I certainly hope the surface area used in the calculation is not just the GC electrode surface area, because greater electrode surface leads to greater current, just as expected.
And of course, how would the current density differ in the case of free porphyrin? In the control as well, care to explain the irreversible wave seen in the nitrogen control?
3) The Koutecky-Levich analysis on the mechanism is a nice touch. In fact, the section on the analysis is very nice, although it doesn’t hurt to spend a little more effort analysis the rather strange number of electron transferred or justifying the reaction is only oxygen reduction.
In the end, this is one of the first step towards MOF-graphene based composite material. Though it has flaws, at least someone somewhere is trying to make the idea work. So, just to throw some project aims out there:
1) Can a framework be constructed where structure binding groups do not interfere with active catalytic centers? (examples in Hupp’s porphyrin systems)
2) Can electron transfer interactions between the backbone graphene and the reaction centers be closer? (reducing electron transfer cost from graphene, or electrode material, to the metal center, when the overpotential is already amazingly low)
3) Can the transformation be catalytic? (since no turnover rate was reported at least for ORR)
4) Can the mechanism involve more than just electron transfer? (i.e., atom transfer chemistry by applying a potential to a pre-treated substrate; not quite catalytic, but could reduce the activation barrier)