
Title: Low Data Drug Discovery with One-Shot Learning
Authors: Han Altae-Tran, Bharath Ramsundar , Aneesh S. Pappu, Vijay Pande
Year: 2017
Journal: ACS Central Science
http://pubs.acs.org/doi/abs/10.1021/acscentsci.6b00367
“Machine learning” may sound to be a “computer-bite” topic, but in today’s chembite, we would discover how machine learning could tell us about a new drug.
Now, imagine a time machine takes you back to the old days, through successes and failures in thousands experiments, you discover some molecules that behave similarly while others do not. Then you probably would look at the similarities among those that look alike, and study their common chemical structures (or functional groups). The next step that you would probably try is to make a new molecule with that chemical structure to see if it also behaves the same. If so, congratulations! You now have a new theory on chemical behaviors.
Time flies, and we are all suddenly back to the present, 2017. With continuous breakthroughs in technologies and computational power, chemists are trying to transform the discovery process by moving from lab onto computer chips. The tool of computation in today’s paper is called a neural network. It can be thought of as a collection of many on-off switches (computer scientists call them nodes). Each of these nodes is activated or deactivated by its neighbours, just like neurons in a human brain. To train a neural network means determining how strongly these switches connect with on another so that it could tackle a task like a human.
The recipes for drug discovery with machine learning are more or less similar as a hands-on discovery. For ingredients, we need data, just like thousands of experiments to be performed in the laboratory. They are the tools to “train” the neural network about which switches are to be on and off. As a result, a “route” is built in the network, analogous to hypothesis of the chemical problems in the old days. Then a molecule new to the neural network is tested. If the network can give the correct prediction, it is said to be recognised (the hypothesis becomes a theory).
The major challenge in computation is to devise accurate predictions to chemical problems without the use of much data. Here, the authors introduce a smart statistical shortcut, called the one-shot learning paradigm, which can significantly lower the amount of data required to construct predictive neural networks. Their one-shot learning maps chemical compounds into a continuous space, where molecules can be particularly to be task-specific. This can even allow the network to choose what to observe from memory.
They also adopted a new deep-learning architecture for the neural network, iterative refinement long short-term memory (LSTM), that could trade information between evidence and query molecules. The iterative refinement can find out the similarities in new experiments, which are related but not the same as those in the training data. It can also perform object recognition, which is crucial for molecular representation. This therefore can significantly improve learning over small-molecules and demonstrated great capability of strong generalisation.
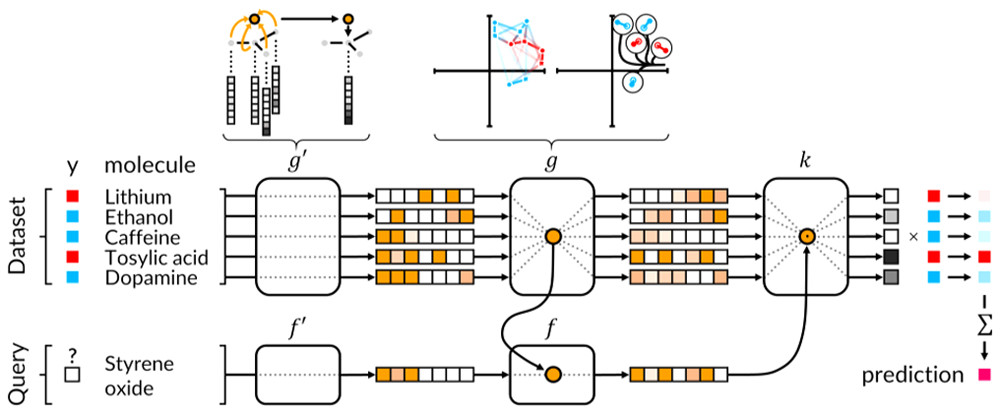
Figure 1 shows how the neural network works. A molecule is given to the network, where its topology and atom features is extracted through some mathematical operations called graph convolutions and pools. With another mathematical operation, the graph gather technique, the features can be recognised and predicted.

Figure 1 Graphical representation of the proposed operations in the neural network. The blue circle represents the switches (nodes) that are in use. The graph convolution and pool are represented for a single node only, but in reality, these nodes operated at the same time.
The authors have demonstrated how chemical prediction can be achieved with small data sets. Computation may still be new to many conventional chemists. Though there are still many challenges and unsolved problems in computational chemistry, it will be interesting to see how crossover between computation and chemistry would bring unprecedented new advances to the world.
Pingback:Less is more – learning chemistry with machine learning – Eli's Science Blog
Pingback:Computational Chemistry > Computers + Chemistry – Chembites