

Title: Crystal Structure Prediction via Deep Learning
Authors: Kevin Ryan, Jeff Lengyel and Michael Shatruk
Year: 2018
Journal: Journal of the American Chemical Society
DOI: 10.1021/jacs.8b03913
Featured image and figures used with permission via ACS AuthorChoice open access
Remember the Magic 8 Ball? The supposed oracle that could provide answers to questions we were unsure about? Recently, scientists have demonstrated the ability to effectively do this with chemical structures.
While there have been great strides in the advancement of chemistry over the last century, we can say we are currently facing a similar challenge with discovering new chemical compounds Over its extensive history, most of the easily synthesized chemical compounds have been discovered. Something that scientists tackle today is the discovery of novel structures and compounds, especially those with complex structures containing elements that can bond in multiple ways in a single structure.
But what if I told you we could predict feasible structures? Structures that have never been discovered? That’s exactly what scientists at Florida State University took on as their newest challenge. Using crystallographic data, which provides a structural representation of an existing compound, they have developed a system based on pattern recognition that can predict potential structures with a degree of accuracy. This is a leap forward from existing methods of structural prediction, which rely heavily on the long standing brute force method of guess-and-check, which is both time-consuming and limited in its scope.
The authors have developed a deep neural network (DNN) utilizing existing crystallographic structures from databases. Within the DNN, there are 118 individual neurons, which each correspond to an existing element. The effectiveness of the DNN is attributed to the non-linear transformations, giving it the edge over existing methods for prediction as it can make the non-linear approximations necessary for prediction involving multiple elements.
They employed 60% of known structures from a crystal database in the learning process of the system, allowing it to identify the various topological and chemical properties of the individual elements. Of the remaining 40%, 20% were used to test the initial model to ensure accuracy and the final 20% were used as a blind test for the final system. When tested with a 2- and 3-element system, the DNN predicted a large number of structures (3051 and 66652 respectively), with all known literature reported structures being included in the output sets.
In Figure 1, we observe a low rate for incorrect classification of elements, where the intensity of color, excluding the diagonal axis, indicates the disagreement between the DNN’s predicted values and crystallographic information from the database. This demonstrates that most of the error displayed by the DNN lies below the 10% threshold. Regions of error correspond to elements with similar chemical properties, a trend that is a “learned” function of the DNN.
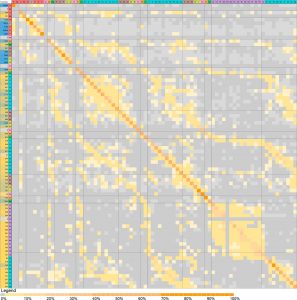
Figure 1. Visual demonstration of accuracy of the DNN in correlating elements that can be assigned in a given position in a structure. Reprinted with permission from Crystal Structure Prediction via Deep Learning, Kevin Ryan, Jeff Lengyel, and Michael Shatruk Journal of the American Chemical Society Article ASAP, DOI: 10.1021/jacs.8b03913. Copyright 2018 American Chemical Society.
While the DNN can predict localized geometry based off topological and chemical information (describing the spatial connectivity and element reactivity) it has learned from existing data sets, a significant drawback with of the system is the lack of negative data sets, or knowledge of what cannot be feasibly formed. The absence of this data results in the DNN being unable to throw out improbable structures. This could be circumvented by the input of a probability function for elements to achieve a specific connectivity. However, this would result in the DNN simply looking up the probability for various combinations, before discarding structures that are deemed unlikely.
The DNN developed by the authors is a huge step towards successful structure prediction. While the method is not yet perfected, it demonstrates immense potential for the discovery of novel structures. With the constant addition of new structural data, the DNN could slingshot the discovery of novel solid-state structures to new heights.