
Title: Reversible Heterolytic Cleavage of the H–H Bond by Molybdenum Complexes: Controlling the Dynamics of Exchange Between Proton and Hydride
Authors: Shaoguang Zhang, Aaron M. Appel and R. Morris Bullock
Year: 2017
Journal: Journal of the American Chemical Society: http://pubs.acs.org/doi/abs/10.1021/jacs.7b03053
Have you ever burned dihydrogen gas, in labs? With the famous ‘squeaky pop’, the combustion of dihydrogen releases vast amounts of energy (Equation 1). The only product of this reaction is water, making H2 a cleaner fuel than hydrocarbons (though NOx pollutants can be produced if N2 is present).
2H2 + O2 → 2H2O ΔfH⊖(298 K) = -286 kJ mol-1 Equation 1
The chemistry research challenges here are to make H2 using renewable energy, store it until it is needed, and then release the energy to do something useful. Today’s paper is focused on the last of those challenges, releasing the energy.
In this work, the authors cleave the H-H bond heterolytically (i.e. forming H+ and H–) using Frustrated Lewis Pairs (FLPs). A reminder that:
A Lewis acid is a species that acts as an acceptor of an electron pair (e.g. BF3 only has 6 valence electrons, but wants 8)
A Lewis base is a species that acts as an electron pair donor (e.g. diethyl ether has an oxygen atom that can donate a lone pair of electrons)
An FLP comprises a Lewis acid and Lewis base at two sites within the same molecule. They are ‘frustrated’ because the pair are prevented from reacting with each other, either by steric hindrance or because the adducts would form highly strained rings. Thus, FLPs contain two very different reactive sites – one that is poised to donate an electron pair, the other primed to accept an electron pair – which is helpful for breaking small molecules apart. The dual functionality of FLPs has become a key tool in small molecule activation and catalysis.
The structure of the FLPs used in this work are shown in Figure 1. The molybdenum centre is acting as the Lewis acid here, whilst the amine group is the Lewis base. As an adduct, the pair form strained four-membered rings that are high in energy, with the amine only weakly coordinated to the metal. The ‘R’ groups allow the steric and electronic properties of the system to be tuned. Interestingly, the authors compare the ‘PNP’ and ‘P2N2’ ligands in Scheme 2 of the paper (i.e.one pendant amine vs two) They hypothesise that the smaller ‘bite’ angle (angle P-Mo-P) of the less flexible P2N2 ligand leads to more strained four-membered rings and weaker interaction between the nitrogen and molybdenum atoms. This would explain why the P2N2 ligand works better – the Lewis acidic and basic sites are not tied up by a strong interaction with each other.
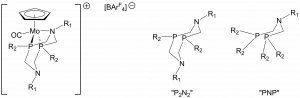
Figure 1 Left: General structure of the H2-splitting FLP compounds studied. Note: this complex is made in situ by hydride abstraction. Right: PNP and P2N2 type ligands.
The weak coordination of the amine allows H2 to coordinate to the metal in its place, forming a complex somewhere on the scale between a dihydride and a H2 ligand (nearer the latter). But now the amine is ‘free’ and can act as a base to deprotonate the bound H2 ligand, leaving behind a metal hydride (Figure 2).
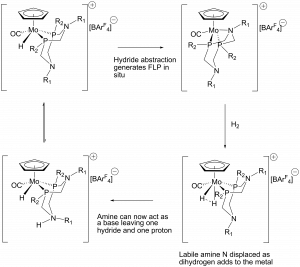
Figure 2: Activation of H2 using a FLP.
How are these compounds characterised? The authors were able to grow crystals of the hydride complexes with a non-coordinating anion. Crystal structures provide precise information about the bond angles and lengths. Here, the authors have used X-ray crystallography to determine the structure, The H-H distance in the structure is too long to be a full bond, whilst the N-H and Mo-H distances are characteristic of a protonated amine and metal hydride. In future studies, neutron diffraction could provide more accurate proton/hydride positions.
Variable temperature NMR spectroscopy, in which NMR spectra of the same sample are recorded at different temperatures, was also revealing – the proton and hydride rapidly exchange at 20ᵒC leading to a broad two-proton signal, but when cooled down to -80 ᵒC, the separate N-H and Mo-H signals become clear. The authors varied the alkyl groups on the phosphines and the pendant amines and measured the rate of exchange for each complex. The trend in exchange rate between the proton and hydride followed exactly the trend in pKa values of the protonated amine species (bottom left of Figure 2). They determined the pKa values by adding base and measuring the ratio of protonated to unprotonated base, which in turn can be used to calculate the equilibrium constant, and therefore the pKa. The lower the pKa, the faster the exchange – Figure 8 of the paper (you can access the paper via the link at the top of the page) shows that plotting the log of the rate constant for exchange against pKa gives a straight line. That is really the take-home message of this paper – a quantitative relationship between exchange rate and pKa that allows us to predict, or tune, the H2 bond-breaking properties.
Why is this important? It helps in designing better catalysts for H2 oxidation. Combine this oxidation catalyst with an O2 reduction catalyst and you get a fuel cell that utilizes dihydrogen – allowing us to use the energy in a controlled way. The benefits of this research extend beyond just H2 oxidation for energy. Hydrogenation is a crucial (and lucrative) chemical transformation that leads to the production of many high-value chemicals. Activating H2 with FLPs, gives a starting point for catalytic hydrogenations, another area under the H2 activation umbrella. The challenge of activating the only bond of the simplest conceivable molecule is worth the frustration.