
Title: Bicarbonate Is Not a General Acid in Au-Catalyzed CO2 Electroreduction
Authors: Anna Wuttig, Youngmin Yoon, Jaeyune Ryu and Yogesh Surendranath
Year: 2017
Journal: Journal of American Chemical Society
10.1021/jacs.7b08345
Reduction or addition of electrons or H equivalents (H++ e–) to a substrate can take place in different ways. Both H+ and e– can be added in the same step (concerted/coupled, known as proton coupled electron transfer/PCET), or electron transfer (ET) and proton transfer (PT) can occur in a stepwise manner. While PCET is thermodynamically more favorable stepwise ET and PT occur when the transfer distance is too long to be travelled by heavier proton. Enzymes use these transfer processes during important natural redox reactions such as oxygen generation by photosynthesis, nitrogen fixation, respiration and DNA synthesis.
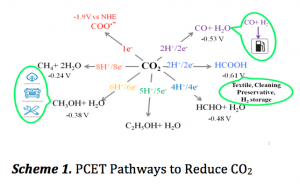
Various PCET pathways imparting different numbers of H+ and e– to carbon dioxide (CO2) give rise to an array of commodity chemicals. As we can see in Scheme 1, products like methanol, carbon monoxide, acetic acid are vastly used in transportation, agriculture, construction, textile industry, cleaning reagents and making preservatives. Thus understanding the rate limiting steps (RLS) and identifying efficient proton donors are desired to enhance the rate of the reactions and to form one product selectively over others.
Electrochemistry is a technique to induce reduction (adding electrons) and oxidation (taking away electrons) by sweeping the voltage/potential from zero to negative and then to the positive direction. This method is particularly useful to have a better control on the rate and precision with which we can add or take away electron. We can reduce CO2 electrochemically to a desired product by adding necessary number of electrons to it at a particular rate in presence of a source of proton/ Lewis acid. Sometimes a much negative voltage/potential is needed to reduce CO2 at a particular electrode (the cathode where the reduction takes place). The difference in potentials at which CO2 is reduced in nature (Eo) and on a particular electrode (Ereact) in known as overpotential (η) and often this parameter measures how good that electrode material or a catalyst is. Changing the material, nature, surface area and shape of the electrode can decrease η. However, there is a trade-off in choosing a material that has lower η with the selectivity of the reaction. For example, a planar polycrystalline gold electrode selectively reduces CO2 to CO, nevertheless, at a very negative potential/ high η in aqueous solution. To make this process more efficient and lower η, it is instrumental to understand the underlying PT and ET mechanism and the corresponding RLS on Au electrode and in the electrolyte solution.
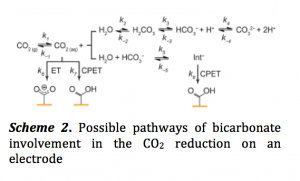
CO2 and water (H2O) forms bicarbonate (HCO3–). While it is known that this HCO3– is not reduced at the electrode surface its exact role is still not established. One possibility is that HCO3– is involved in the acid-base equilibrium. It equilibrates with CO2 to buffer the pH of the electrolyte. Since the rate of electrochemical reduction is slower than this equilibrium, the backward reaction from HCO3– to CO2 is feasible. Another possibility is the involvement of HCO3– as an acid to donate protons to the CO2 adsorbed on the electrode surface (Scheme 2).
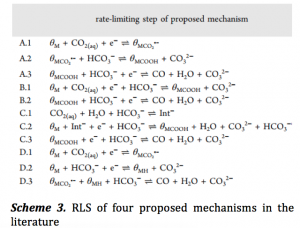
The present work aims at finding the exact role of bicarbonate using kinetics and modeling experiments. The authors considered four widely accepted mechanisms for CO generation on Au electrode involving HCO3– as the proton donor (Scheme 3). The sequence of PT and ET, or PCET is different in these four mechanisms. The order (ρ) of these electrochemical reductions are calculated in terms of the change in CO production rate with respect to the change in concentration of HCO3– ([HCO3–]). With the generation of CO we should observe a change in the current density (jCO) and so ρ is formulated as –
ρ= (δlogjCO/δlog[HCO3–])……………….(1)
Conventionally ρ is measured at constant η (Eo– Ereact). However, the authors in this work pointed out that some error can be introduced in this way of estimating η since jCO changes with Eo. And Eo also changes with the change in concentration of the CO2 and CO on the electrode surface. As Au is known to reduce CO2 at a much negative potential, far away from Eo, the authors measured the order (ρ) at a constant Ereact. They carefully chose three potentials at which the reduction is not dependent on how fast CO2 is supplied to the electrode so that ρ is a function of jCO and [HCO3–] only and no other variables.
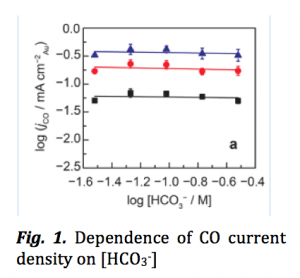 Fig. 1 shows that jCO is independent (or has zero-order dependence) of [HCO3–] in the bulk electrolyte. jCO is also insensitive of the pH (= -log10[H+])change that happens when [HCO3–] increases. Their result is contrary to the findings by a previous work where a 0.9 order in [HCO3–] was found. This disparity is believed to be caused by the presence of phosphate that was used as a co-electrolyte in that work. The authors showed that the phosphate indeed inhibits the CO formation on Au surface. And to accommodate for this inhibition, the order needs to be adjusted to 0.9 with respect to [HCO3–] in presence of phosphate and in its absence the true order for [HCO3–] is essentially zero.
Fig. 1 shows that jCO is independent (or has zero-order dependence) of [HCO3–] in the bulk electrolyte. jCO is also insensitive of the pH (= -log10[H+])change that happens when [HCO3–] increases. Their result is contrary to the findings by a previous work where a 0.9 order in [HCO3–] was found. This disparity is believed to be caused by the presence of phosphate that was used as a co-electrolyte in that work. The authors showed that the phosphate indeed inhibits the CO formation on Au surface. And to accommodate for this inhibition, the order needs to be adjusted to 0.9 with respect to [HCO3–] in presence of phosphate and in its absence the true order for [HCO3–] is essentially zero.
A modeling study is additionally performed to compare the effect of diminishing concentration of HCO3– near the electrode surface owing to the fast consumption of H+ and sluggish formation of HCO3–. Their model illustrates that jCO is independent of local [HCO3–] when the ET mechanism is in action (mechanism A in Scheme 3) and they are inter-dependent when the PCET mechanism (B) takes place.
Both the experimental and modeling studies indicate that mechanism A or D to be the most plausible ones where the rates are independent (or of zero-order) of [HCO3–] and pH, and first-order (rate is proportional to [CO2]) with respect to CO2. The only difference between mechanism A and D is the step at which HCO3– adds H+. Since this step happens after RLS it does not affect the order. This work also unequivocally rules out the involvement of hydronium (H3O+) or water as the proton donor in RLS on Au electrode as indicated by previous studies. While this work involves a thorough and well grounded study onCO2 reduction on Au electrode, it can be followed to understand other electrodes as well to better promote electrochemical CO2 reduction via low barrier PCET instead of ET as we see in case of Au.