
Title: Self Healing Catalysis in Water
Authors: Cyrille Costentin and Daniel G. Nocera
Year: 2017
Journal: Proceedings of the National Academy of Sciences
During today’s global energy crisis, the sun is the only source of abundant and carbon-free energy. Plants use photosynthesis to harness the solar energy into carbohydrates and oxidize water (H2O) to generate diatomic oxygen (O2) at the same time. Therefore, nature is our inspiration in making synthetic molecules to split H2O.
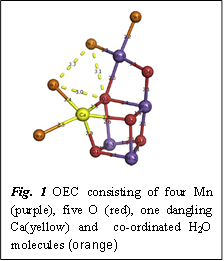
In all known organisms that are capable of carbon assimilation through photosynthesis, oxygen evolution takes place inside a specialized protein named Photosystem II (PSII) in presence of light. Inside PSII there exists an inorganic oxide molecule known as the Oxygen Evolving Complex (OEC) which is primarily responsible for the rearrangement of bonds between two H2O molecules to form O2. The OEC consists of four manganese atoms connected by bridging oxygen atoms and a calcium atom (Fig. 1).
 The OEC is a highly optimized system and each mole of it can form as much as four hundred moles of O2 per second under ambient conditions. However, the OEC is not very stable in the presence of high concentrations of O2 and solar UV radiation. Plants need to replace the OEC twice every hour to keep up the efficiency of O2 evolution. Although the instability of OEC in nature is unimportant as it can be replenished naturally, the stability for synthetic molecules for H2O splitting over time, is of great concern to the scientists. Most molecules (or catalysts) lose their activity after a finite number of reaction cycles. Achieving higher efficiency with improved stability is very desirable for any catalyst.
The OEC is a highly optimized system and each mole of it can form as much as four hundred moles of O2 per second under ambient conditions. However, the OEC is not very stable in the presence of high concentrations of O2 and solar UV radiation. Plants need to replace the OEC twice every hour to keep up the efficiency of O2 evolution. Although the instability of OEC in nature is unimportant as it can be replenished naturally, the stability for synthetic molecules for H2O splitting over time, is of great concern to the scientists. Most molecules (or catalysts) lose their activity after a finite number of reaction cycles. Achieving higher efficiency with improved stability is very desirable for any catalyst.
In artificial photosynthesis the goal is to convert sunlight into spatially separated electron/hole pairs within a photovoltaic cell and then to capture these charges with catalysts that mediate water splitting. The four holes are captured by a catalyst at the anode to produce oxygen at a less positive potential (more thermodynamically favorable), and the four electrons are captured by a separate catalyst at the cathode to produce hydrogen at a less negative potential (Scheme 1). The net result is the storage of solar energy in the chemical bonds of H2 and O2.
Several metal oxides are routinely used for oxygen evolution from basic and acidic aqueous solution. Interestingly at pH=7 these metal oxides are the most basic species present in solution, so they react with the protons, generated from water oxidation. In this way they slowly get dissolved into the aqueous solution. Therefore, developing stable catalysts to perform the water oxidation at neutral pH, and at less positive potential is highly desirable, yet challenging.
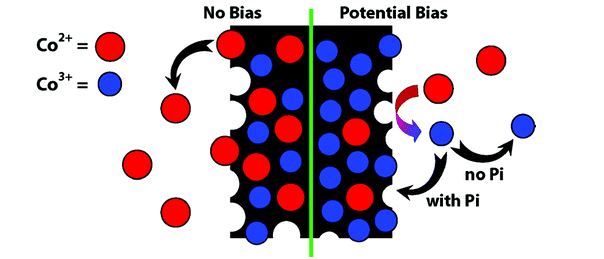
An important breakthrough in this field was made by Daniel G. Nocera and his students at MIT in 2008. They noticed that when they mixed any cobalt salt with a phosphate buffer in aqueous solution, a cobalt (II) oxide film was being deposited on the electrode. They further showed that this material was highly active for water oxidation since it continuously repaired itself automatically after decomposing at end of each reaction cycle. While the discovery of this self-healing catalyst was serendipitous and the exact chemical formula of the catalyst was not known, they established that it was composed of cobalt and phosphorus (CoPi) from here on. Since then a number of follow-up studies have been done on this CoPi OEC to better understand the oxidation state of the intermediate Co species mainly by X-ray Absorption Spectroscopy (XAS) and Electron Paramagnetic Resonance (EPR) spectroscopy.
In a more recent work, Costentin and Nocera discussed why this self-healing mechanism works for their catalyst, as well as how to rationally design other self-healing water oxidation catalysts by better understanding the kinetics of the key catalytic steps. The catalytic cycle has been postulated from experimental results as shown below (Scheme 2).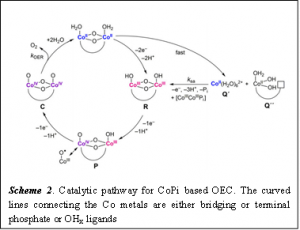
We can see that the cycle involves Co(II), Co(III) and Co(IV) oxo species. To compare the reactivity of these Co species of different oxidation state, we have to understand their electronic configuration a little bit. Since Co is a transition metal it has 9 electrons in the d orbital (remember, s, p and d orbitals can have maximum 2, 6 and 10 electrons respectively). The d orbitals split in two different energy states in inorganic complexes as shown in Fig. 2, a phenomenon known as Crystal field Splitting. For Co(II) and Co(III) the electrons are arranged in the way shown in Fig. 2. Since there is a larger number of unpaired electrons present in Co(II) than in Co(III) it is easier for those unpaired electrons in Co(II) to react . Hence Co(II) is considered as reactive while Co(III) is inert.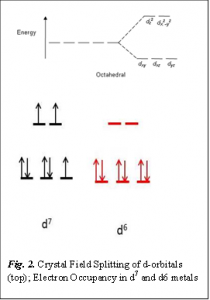
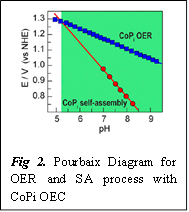
In aqueous solution Co(II) reacts with H2O and gets hydrolyzed fast to form Q’ and Q” (Scheme 2). However, these two species can quickly self-assemble (repair) to form R which is comparatively inert/stable Co(III) species. Now to understand why this self-assembly process is so efficient, we have to understand the kinetics of this step. If we look at Scheme 2, it is evident that the step of self assembly (SA) releases three equivalents of protons, so the rate of this step should have an inverse third order dependence on the concentration of proton (rateSA= kSA*[H+]-3). R is a stable Co(III) species, so to convert R into P is the slowest step in the overall catalytic cycle (or, rate determining step). As this step involves release of one equivalent of proton, the rateOER= kOER*[H+]-1. For enabling an efficient self-assembly process, its rate needs to be faster than the OER at pH=7. That’s what the authors have shown by a series of thermodynamic equations. Mathematically the rate of a reaction correlates to the reduction potential (E) of the reactions. The Nernst equation relates pH (-log10[H+])to E (Scheme 3). This relationship between the pH and E is graphically shown in a Pourbaix diagram.
Comparing the rates of the OER and self-assembly, it can be clearly understood that with the increase in pH, the rate and the reduction potential of the former (related to [H+]-1) will decrease less compared to the latter (related to [H+]-3). Due to this difference in rate, in Fig. 3 we can see that in the green-zone where the pH range is 5.2-9.5, the self assembly can occur faster and at a lower potential than the OER. This work also suggests other strategies like optimizing the cell volume and electrode surface area to achieve better self-repair mechanism.
This work is very relevant in advancing our understanding of the present self-healing OEC and furthering our search for similar catalysts with judicious choice of other metal and buffer combinations. Use of cheap and abundant metal catalysts that can repair themselves and can be efficient for long periods of time, is highly attractive given the present global energy demand.