
Title: Electrocatalytic Radical Dichlorination of Alkenes with Nucleophilic Chlorine Sources
Authors: Niankai Fu, Gregory S. Sauer, and Song Lin
Journal: Journal of the American Chemical Society
http://pubs.acs.org/doi/10.1021/jacs.7b09388
Year: 2017
The unsaturated bond of an alkene is one of the most exploited functionalities inside the organic chemist’s arsenal, and for very good reason! This double bond lends itself to a palette of chemical reactions such as hydrogenation across the double bond to get the reduced alkane, hydroformylation to generate aldehydes, polymerization processes that yield industrially useful polymeric materials, or oxidative difunctionalizations to introduce two vicinal heteroatoms (Figure 1).

Figure 1: Representative reactions of alkenes
Oxidative difunctionalizations in particular are a linchpin of alkene transformations because they allow chemists to not only affix two heteroatoms (e.g. O, Cl, Br, I) across the double bond but also because, if employed under appropriate catalytic conditions, they enable the installation of these heteroatoms in a stereoselective fashion, favoring their incorporation in one spatial arrangement over all other possible spatial combinations. Unlike stereoselective alkene dihydroxylations, which have enjoyed a well-established success, stereocontrolled alkene dihalogenations protocols are relatively in their infancy. Yet, the prevalence of halogens at stereocenters in important natural products and drugs underscores the need to develop reliable stereoselective variants for this chemical maneuver.
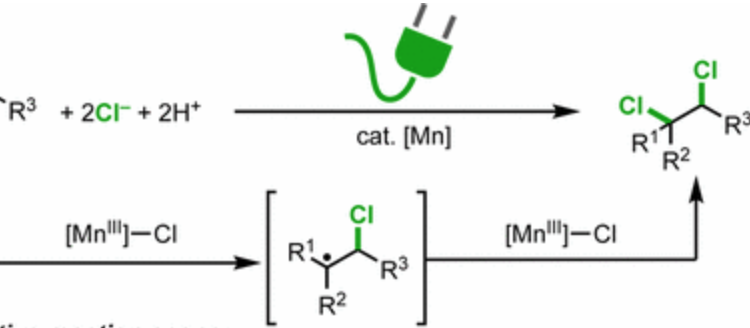
Some successful attempts do exist already, such as selenium-catalyzed dichlorinations or dichlorinations with electrophilic sources of chlorine, generated in situ by combining reactive chlorinating reagents with an oxidant (Figure 2A, 2B). these approaches, however, require stoichiometric rather than catalytic amounts of oxidants, reducing compatibility with many functional groups and generating large amounts of harmful waste. In the current account, the lab of Song Lin at Cornell University introduces an attractive alternative by combining electrocatalysis and transition-metal catalysis for the stereoselective dichlorination of alkenes under very mild conditions (Figure 2C). The success behind electrocatalysis lies on the fact that the application of enough electrical potential can cause organic substrates to loose (get oxidized) or gain electrons (get reduced) in a predictable fashion and without the use of reactive activating agents used commonly in organic synthesis, thus providing an efficient and suitable complement to more traditional chemical methods.

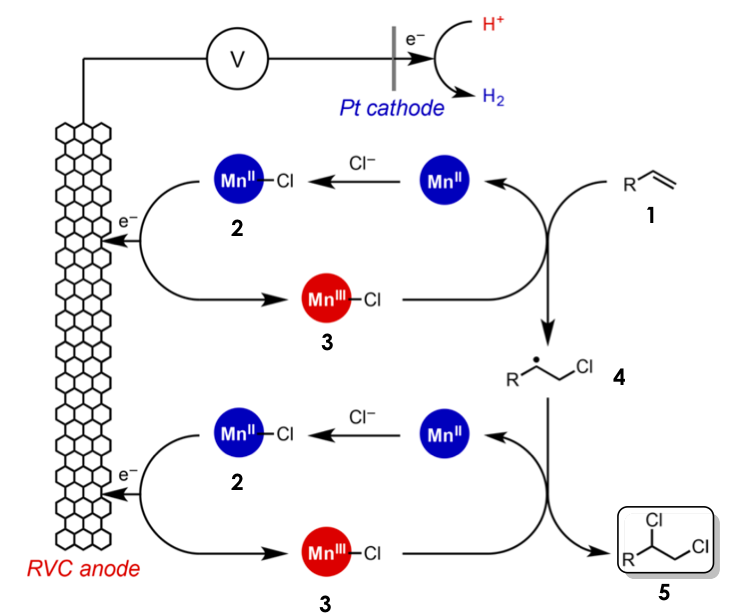
Lin and coworkers envisioned that the use of a lithium perchlorate (LiOCl4) electrode in combination with a redox-active metal such as manganese II (MnIII) could facilitate the activation of an alkene towards dichlorination with magnesium chloride (MgCl2) as the source (Figure 3). More specifically, the anionic chloride forms a complex with MnII (2), which undergoes a one-electron oxidation when an electrical potential is applied at the anode to form a MnIII chloride-transfer complex (3). This species now has oxidizing properties and will abstract an electron from the substrate (1) while donating one equivalent of halogen to the less substituted side of the alkene to generate the more stable a-halo radical (4). A second round of this cycle adds the second equivalent of halogen to afford the vicinal dichloride product (5) with an anti- configuration, meaning that both chlorides were incorporated from opposite faces of the pre-existing alkene Finally, the electrons harvested by the anode are transferred to the cathode and finally to acetic acid, which acts as the final sacrificial oxidant, generating magnesium acetate and hydrogen gas as the sole byproducts
This strategy The authors attribute the observed stereoselectivity to the relatively large steric profile (size) of the MnIII-Cl transfer agent (3) and the fact that this chloride also bears a relatively large partial negative charge. Both of these properties deter the second equivalent of chloride from adding on the same side of the previously installed halogen, thus yielding the anti– stereoisomer.
The protocol worked for a wide palette of substrates, including cyclic alkenes, styrenes, and mono-substituted, di-substituted, and trisubstituted aliphatic (acyclic) alkenes in excellent yields. Additionally, the relatively mild radical-generation nature of this system was compatible with functional groups that are highly sensitive to oxidative conditions such as alcohols, aldehydes, amines, sulfides, and N-containing heterocycles. These moieties would not survive more traditional dichlorination protocols which rely on strong oxidants to generate electrophilic chloride, yet they remained intact in Lin’s method. Finally, the strategy was also highly scalable to multi-gram preparations and required relatively trivial purification steps to obtain spectroscopically pure product. To find out more about the optimization process and the intricacies of substrate scope, the curious reader can search through the article itself.
In short, Lin’s group successfully combined redox-based transition metal catalysis and electrocatalysis to develop an elegant approach for the anti- dichlorination of alkenes. The system’s generous substrate tolerance, high yields, and mild nature make it a strong contender in the long-sought path to find robust alkene halofunctionalizations. Although more detailed mechanistic information, particularly for the stereo-defining step, would be interesting to have at the moment, one cannot help but praise the development of this novel reactive manifold and hope that this effort will encourage more gallant immersions in the field of alkene dehalogenation.