
Title: Selective Radical Trifluoromethylation of Native Residues in Proteins
Authors: Mateusz Imiolek, Gogulan Karunanithy, Wai-Lung Ng, Andrew J. Baldwin, Véronique Gouverneur, Benjamin G. Davis
Journal: Journal of the American Chemical Society
Year: 2018
The desire to tailor biology’s building blocks has always been an ambitious goal for synthetic and biological chemists. While it is true that Nature can perform astounding chemical reactions in a fraction of the time than the most adroit chemist and orders of magnitude more selectively than the most revered synthetic transformation, its existent chemical diversity can sometimes fall short to the ever-growing curiosity of scientists.
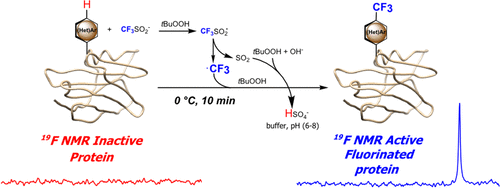
Adding novel functionalities to the chemical language of cells can help us exploit their already prodigious synthetic abilities or can allow us to study their components at a molecular level that was unprecedented just a few decades ago. For example, site-specifically affixing a very chemically distinct functional handle to a protein opens the possibility to interrogate such protein selectively amongst its complex biological milieu. In a new account published in JACS, the lab of Ben Davis at the University of Oxford elegantly illustrates this notion. They present a new strategy to directly add fluorine-containing groups to proteins after these have been translated, or assembled, by the cell. To appreciate the impact of this research, it is first necessary to understand the motivation that led the group towards their discovery (Figure 1).
Fluorine atoms are absent from nearly all natural products and primary metabolites made by cells; nonetheless, their use is widespread in medicinal chemistry. In the world of pharma, fluorine is a favorite since its addition to small molecules such as drug candidates drastically alter their solubility and their interactions with proteins. For clinical diagnostics, incorporation of a radioactive isotope of fluorine, 18F, into a pharmaceutical allows observing a specific metabolic process via PET imaging by virtue of where is that radiopharmaceutical compound broken down inside a patient’s body. It comes as no surprise, then, that the incorporation of fluorine into proteins could open avenues to study how proteins interact with each other or to develop fluorine-based imaging techniques for native proteins, such as 18F PET imaging or 19F NMR spectroscopy.
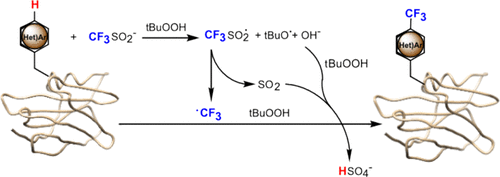
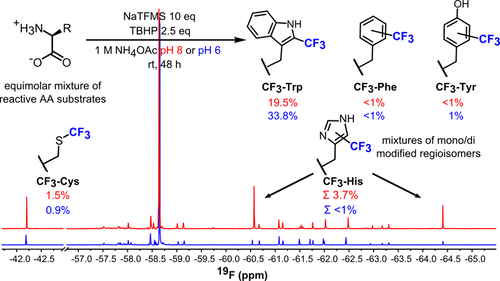
The Davis group started by considering the benign nature of carbon-based radical precursors to form C-C bonds with unactivated C-H bonds in proteins (radicals are chemical species with an unpaired electron). With this in mind, they searched for regents that would produce a trifluoromethyl radical, herein written as ·CF3, to functionalize the side chains of several amino acid monomers. A quick 19F NMR analysis of the reaction revealed that sodium trifluoromethylsulfinate (NaTFMS) could trifluoromethylate the side chains of aromatic amino acids (tryptophan, tyrosine, phenylalanine, and histidine) and of sulfur-containing cysteine. Further tweaking of the reaction conditions allowed for a ~30-fold more selective functionalization of the C2 of tryptophan over all the other amino acids (Figure 2). The curious reader can refer to the original paper to explore more.
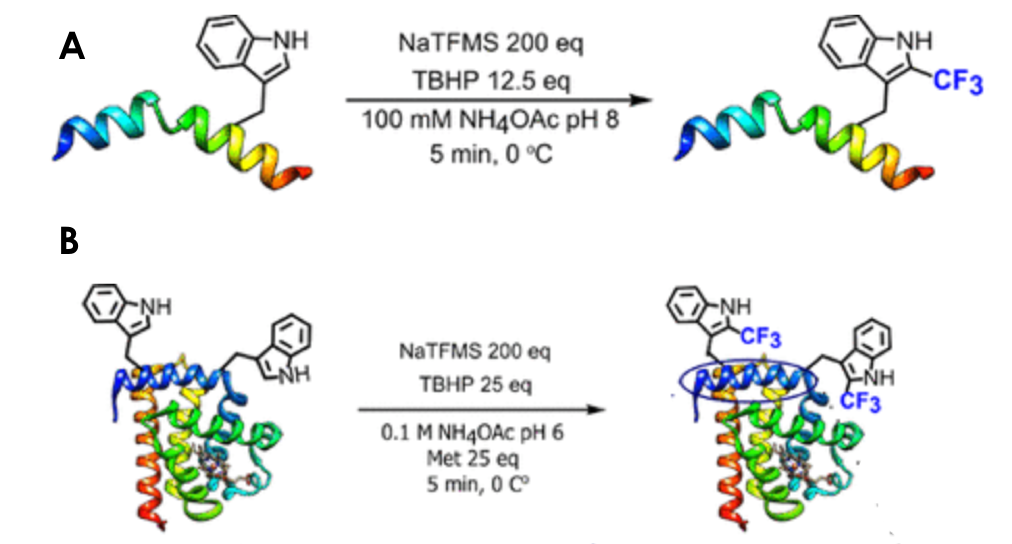
To investigate this approach on a more complex substrate, the NaTFMS reaction was carried out on melittin, a small 26-residue peptide with a single tryptophan. Analysis of the product by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) revealed successful trifluoromethylation at the unique tryptophan (Figure 3A). More challenging substrates such as horse hear myoglobin (2 tryptophan residues) and lysozyme (6 tryptophan residues) were also probed in this fashion and generated the expected trifluoromethylated species with the respective number of functionalized tryptophans even in the presence of abundant cysteines, thus showcasing the selective nature of this strategy (Figure 3B). Moreover, the radical character of this transformation was confirmed by the addition of radical “scavenger” such as TEMPO, which quenched the ·CF3 species and arrested the labeling reaction.
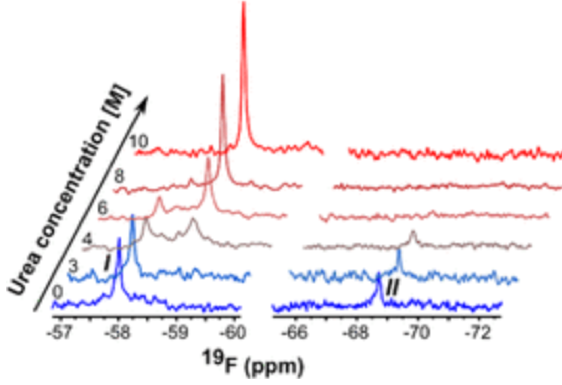
The method’s validity was finally probed via 19F NMR spectroscopy as a proof-of-concept. The trifluoromethylated horse heart myoglobin was analyzed via this methodology to reveal two expected signals in the 19F spectrum corresponding to the two different trifluoromethyl tryptophans in different environments. Destruction of secondary and tertiary structure by thermal denaturation or chemical denaturation with high concentrations of urea promoted the coalescence of these signals into a single peak as the tryptophans’ distinct local environments were perturbed. Similarly, digestion of the protein into single monomers collapsed the two signals into one (Figure 4).
Overall, however, the strategy presented by the Davis group sells as quite attractive as it provides residue-specific labeling of complex proteins under relatively mild conditions and fast (~10 min) reaction times. There are, of course, several questions that remained unanswered and a variety of follow-up that needs to be performed to implement this technology in live cells. It would be interesting to get a mechanistic explanation for the robust selectivity of this reaction, particularly on why tryptophan is functionalized at C2 since it is usually the C3 or C7 positions the ones that show higher reactivity in enzymatic functionalization reactions. A more pressing question is perhaps the applicability of this methodology in mixtures of proteins. It is evident that the current conditions will not selectively functionalize one tryptophan-containing protein over another, but it would be interesting to see how the group pursues this research such that it could be applied to cellular lysates or even live cells.