

Featured image: Adapted from inspiredbythemuse/pixabay
The recent Harry Potter fanfiction is a wonderful mimic of J.K. Rowling’s distinctive style. It’s whimsical, high-stakes, and filled with the playful banter of our favorite magical trio. But with lines like “Ron’s Ron shirt was just as bad as Ron himself,” and a plot that jumps around like a strange dream, any human reader will immediately know that something is up.
That’s because it was written by a computer. Or rather, a type of artificial intelligence (AI) “trained” on the seven-book series and made into a predictive text keyboard, like the one on your smart phone. And, admittedly, with a little editorial help from humans as well.
While this is not the fully functioning humanoid robot that we usually think of as AI, it is our robot overlord’s predecessor: computers that can learn, formally known as “machine learning.” But long before we were teaching computers to write Harry Potter and the Portrait of What Looked Like a Large Pile of Ash, we were teaching them how to do something even more important: chemistry.

The first paragraph of Harry Potter fanfiction, generated in collaboration with AI, from Botnik Studios.
Making molecules is perhaps what chemists are known for best, but much like this Harry Potter fanfic, some chemists have begun collaborating with computers. Machine learning eliminates the toil of trial-and-error from the synthesis lab, making it a much happier, more productive place. It is being used in the labs of Harvard, MIT, and Stanford to predict chemical reactions, discover new drugs, and more recently, optimize reaction conditions in practically no time.
Researchers in the Zare lab at Stanford were able to optimize four chemical reactions with a type of machine learning called “deep reinforcement learning.” This wasn’t just a simulation, either – the reactions were done in microdroplet form, observed by the computer using mass spectrometry, and the conditions were adjusted by the computer based on previous results. With this method, the chemical reactions were optimized in just 30 min, and used 70% fewer trials than state-of-the-art optimization algorithms.
While this may sound mundane, make no mistake, it is a big deal. Why? The pharmaceutical industry. This huge, multi-billion dollar industry rests its foundation on the chemist’s ability to create molecules, either dreamed up on a chalk board or inspired by nature, with extreme precision.
Unfortunately, organic chemistry is inherently messy and difficult to control. Even after the “recipe” for making a molecule has been established, optimizing it is time-consuming, human-error prone, and frankly boring. And while most chemists think of optimizing a reaction for yield, pharmaceuticals may want to be optimized for purity or lowest cost instead. Thus, optimization can save millions of dollars in initial materials and provide more effective drugs, potentially eliminating side-effects caused by impurities.
The general method for optimization with the Zare lab’s “Deep Reaction Optimizer” (DRO) is similar to what any chemist would do: change one reaction condition while fixing all others, measure the reaction result, and use this result along with previous trials to inform which condition is changed next. In computer-speak, this is known as reinforcement learning: a “decision-maker” takes sequential “actions” to maximize a cumulative “reward.” In this case, the reward is the optimized reaction, which can be programmed for yield, purity, or cost.
The big question is how does the DRO determine which “actions” will lead to the optimized reaction? Or, rather, how does it actually learn? For this, the researchers used a neural network – a type of machine learning named after the way it mimics the neural connections within the human brain to acquire knowledge through experience. For each new trial of the reaction, the neural network is given the previous reaction condition and reaction result, which are added to the neural network’s memory. The neural network decides the next reaction condition, and selects which memory, or “history,” to pass forward. It is this history information that allows the neural network to interpret the effect of reaction condition on reaction result, and then make a decision for the next trial.
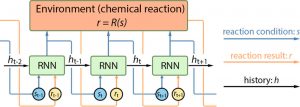
Scheme 1. Visualization of the Deep Reaction Optomizer (DRO) and the recurrent neural network (RNN). From Zhou et. al.
Just like humans, a neural network improves its performance through training. While actual chemical reactions were done for the final tests of the DRO, it was initially trained in simulations. These simulations were not reactions at all, but functions with many local minima and maxima that challenge the DRO to find the global maximum – the very tallest peak in a sea of mountains. When applied to a chemical reaction, the global maximum represents the highest yield, highest purity, or lowest cost, which are each a function of reaction conditions.
After training, the DRO was challenged with 5000 random functions. Averaged over all of these functions, the DRO was able to minimize average “regret,” a measure of distance from the global maximum, in significantly fewer steps than other methods (Figure 1). With this boost of confidence, the DRO was ready to face the real world – or at least the chemistry lab.
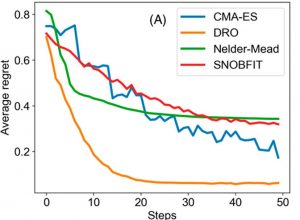
Figure 1. The DRO reaches the global maximum of a function, minimizing “regret,” faster than state-of-the-art algorithms (CMA-ES, Nelder-Mead, SNOBFIT). From Zhou et. al.
Remember when Harry Potter became inexplicably good at potions after following the instructions in the Half-Blood Prince’s potions book? That’s pretty much what the DRO does for the average chemist – you can have all the best algorithms you like, and even Hermione, but the DRO finds shortcuts you didn’t know existed. All four reactions optimized for product yield by the DRO were completed in ~30 steps, rather than the 100 or more steps taken by other methods (CMA-ES and OVAT), despite these reactions being quite different (Figure 2).
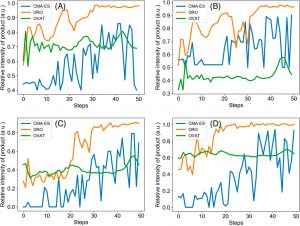
Figure 2. Comparison of optimization methods for different chemical reactions. DRO (yellow) outperforms state-of-the-art algorithms CMA-ES (blue) and OVAT (green) every time. (A) Pomeranz−Fritsch synthesis of isoquinoline (B) Friedländer synthesis of a substituted quinolone (C) Synthesis of ribose phosphate (D) Reaction between 2,6-dichlorophenolindophenol and ascorbic acid. From Zhou et. al.
The DRO’s ability to learn quickly was pretty remarkable. In fact, when the DRO optimized an actual chemical reaction, like in Figure 2 above, it was able to optimize more quickly if it had previously optimized a different chemical reaction. This resulted in the DRO needing 20-30 fewer steps to optimize a reaction.
Much like magic, machine learning is a powerful tool in the chemistry lab, where it casts a cloak of invisibility over the trial-and-error required to optimize and predict reactions, and can even conjure up promising new drugs and how to make them. While I love Harry Potter more than almost anyone I know, the science of computers making medicines cheaper and more effective is even more enchanting.
Paper: Optimizing Chemical Reactions with Deep Reinforcement Learning, ACS Central Science, Dec 2017
Authors: Zhenpeng Zhou, Xiaocheng Li, and Richard N. Zare
This is an unofficial adaptation of an article that appeared in an ACS publication. ACS has not endorsed the content of this adaptation or the context of its use.
—————————————————————————————————————————–
P.S. Here are more of my favorite examples of what machine learning and neural networks can do.
Image Synthesis From Text With Deep Learning (YouTube Video)
All of these faces are fake celebrities spawned by AI
SunSpring | A Sci-Fi Short Film – screenplay written by AI