
Image credit: National Cancer Institute
All figures reprinted (adapted) with permission from “Reversibly Switchable, pH-Dependent Peptide Ligand Binding via 3,5-Diiodotyrosine Substitutions” Chayanon Ngambenjawong, Meilyn Sylvestre, Heather H. Gustafson, Julio Marco B. Pineda, and Suzie H. Pun.ACS Chemical Biology 2018 13 (4), 995-1002. Copyright 2018 American Chemical Society.
Title: Reversibly Switchable, pH-Dependent Peptide Ligand Binding via 3,5-Diiodotyrosine Substitutions
Authors: C. Ngambenjawong, M. Sylvestre, H.H. Gustafson, J.M.B. Pineda, S.H. Pun
Year: 2018
Journal: ACS Chemical Biology
https://pubs.acs.org/doi/10.1021/acschembio.8b00171
Cancer is one of the most prevalent diseases in the United States, expected to affect 1.7 million new patients in the U.S. this year alone. Despite its prevalence, there is no one-size-fits-all treatment for cancer due to its large genetic variability. However, recent advances in cancer therapeutics have capitalized upon common elements in some tumor types, allowing for targeted drug delivery. These treatments can avoid many of the harmful side effects that accompany broad-spectrum chemotherapy by pinpointing malignant cells using features on the cell surface as well as environmental cues.
One therapy involves the use of “prodrugs”, molecules that are in a masked (inactive) form until they are triggered by a external signal. These signals can include the activity of a particular enzyme or a slight change in pH. Many of these prodrugs offer improved target specificity since their triggers are exclusively within the small area (microenvironment) surrounding tumor cells. Prodrugs can achieve similar (or better) tumor-killing rates compared to traditional chemotherapy and require lower dosage because they have such high specificity. However, although prodrugs selectively turn ON in a microenvironment, there is no guarantee that the ON form of the drug cannot seep into surrounding tissue and cause other harmful side effects. The authors of this paper sought to address this problem by designing a system with the capability to alternate between ON / OFF states in the corresponding microenvironment. This kind of reversible prodrug therapy could further decrease drug dosage and create fewer side effects in healthy tissue.
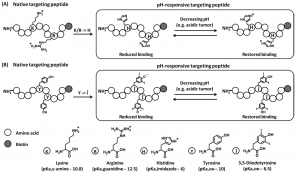
Figure 1: Theoretical illustration of tumor microenvironment (low pH)-responsive targeting peptide engineering via (A) histidine and (B) 3,5-diiodotyrosine substitutions.
To obtain a pH-sensitive, reversible drug delivery system, the researchers first started with a peptide sequence (M2pep) that targets M2 macrophages. These cells are known to congregate in low pH (acidic) tumor regions with low oxygen content. They hypothesized that they could modify key amino acids on the peptide and add pH-responsive histidine (W) and 3,5-diiodotyrosine (Î) residues to promote activity in low pH environments. The idea is that both of these inserted residues are protonated at pH 6 (which is a pH typical of a tumor microenvironment) and this additional proton can interact with key negative charges on the peptide’s paired receptor. Once the peptide leaves the tumor microenvironment and is exposed to tissue at pH 7.4 (normal cellular pH), the peptides are deprotonated and left with a net negative charge. Because ‘like repels like’ for electric charge, these peptides can no longer form a charge-based interaction and will not bind to the receptor (Figure 1). This mechanism allows for a very cleverly designed ON-OFF switch!
The researchers were able to tweak this switch by modifying the peptide sequence and changing the pattern of pH-sensitive residues within regions that directly interact with the cell receptors. They have shown that alternating amino acids histidine (W), tyrosine (Y), and 3,5-diiodotyrosine (Î) can have markedly different effects on an acetylated form of the peptide (AcM2pep(RY)Biotin).
The binding strength of the peptide was tested by adding a dye that would increase in brightness if the AcM2pep(RY)Biotin bound to receptors on the M2 macrophages. In Figure 2, one can see that the brightness of the peptide changed drastically at pH 6 versus pH 7.4 depending on the sequence of residues. The effect is even more pronounced if multiple H and Î amino acids are added (Figure 2b).
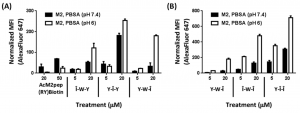
Figure 2: Binding of (A) monosubstituted and (B) disubstituted peptide analogues with M2 macrophages at pH 7.4 and 6.
The best peptide sequence (dubbed Y-Î-Î or Ac-Y-Î-Î if acetylated) was shown to bind selectively to M2 macrophages over another, similar cell line (M1). This is an encouraging result because it means that the peptide can distinguish between tumor (M2) and normal (M1) tissue in this model. It was also shown to be stable over a 48-hour period, outlasting some of the earlier peptide designs thanks to the mutant residues and acetyl group (Figure 3).
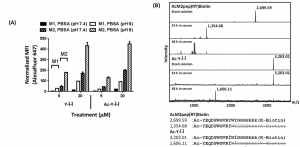
Figure 3: (A) Binding of Y-Î-Î and Ac-Y-Î-Î with M1 and M2 macrophages at pH 7.4 and 6. (B) Stability of modified (Ac-Y-Î-Î) and unmodified (AcM2pep(RY)Biotin) in normal mouse serum.
Although these results are still very preliminary, they are quite exciting! Though modest, there is a clear switching mechanism between the the two pH levels and the peptide sequence appears to selectively target a single cell receptor. This technique may pave the way for other switchable drug targeting systems that can reduce the harmful side effects of many cancer therapeutics.