
Featured Image Source: https://www.genengnews.com/news/removal-of-damaged-mitochondria-helps-treat-chronic-inflammatory-disease/
Primary Source Info:
Title: Target Discovery of Selective Non-Small-Cell Lung Cancer Toxins Reveals Inhibitors of Mitochondrial Complex I
Authors: Nikhil Madhusudhan, Bin Hu, Prashant Mishra, Josè F. Calva Moreno, Khushbu Patel, Richard Boriack, Joseph M. Ready, Deepak Nijhawan
Year: 2020
Journal: ACS Chemical Biology
DOI: http://dx.doi.org/10.1021/acschembio.9b00734
Traditional cancer treatments are frequently associated with severe side-effects and poor treatment outcomes for patients. While sometimes effective, surgery, radiation, and chemotherapy are all nonspecific anticancer therapies, meaning they all have the capacity to significantly damage healthy tissue. On the other hand, targeted cancer therapy differs from nonspecific therapy in that it specifically targets elements of cancer’s biology that fuel tumor growth and that distinguish it from healthy tissue. In many cases, these drugs are more effective and have fewer side effects than nonspecific therapies. One commonly used category of targeted therapies is the small molecule receptor tyrosine kinase inhibitors (RTKis), which act on proteins that cancer uses to bypass normal regulation of cell growth (Fig. 1).
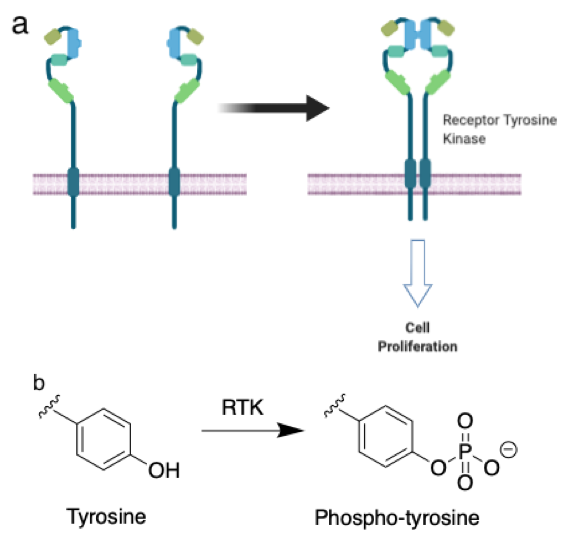
Figure 1: a. Receptor tyrosine kinases are membrane proteins that sense extracellular molecules and transduce this signal to the cytosol. In their inactive form (left), RTKs do not transmit signals to the interior, but upon activation (right) can initiate “programs” that lead to changes in cell behavior. b. RTKs add phosphate groups to tyrosine residues in order to transmit signals. –Justin Donnelly
Cancer cells often have genomes that are heavily altered (mutated) compared to healthy tissue, but scientists have come to understand that a small portion of these alterations, which are called “driver mutations”, are responsible for “driving” the advancement of the disease. Many driver mutations are found in genes for receptor tyrosine kinases (RTKs), signaling proteins in a cell’s plasma membrane which bind extracellular factors and transduce this information into the cell interior by adding phosphate groups to their tyrosine amino acid residues. Blocking an RTK’s ability to phosphorylate its tyrosine residues with an RTKi can choke off the pro-growth signaling that a cancer cell is addicted to, leading to cell death (Fig. 1). Another consequence of the abundance of mutations in cancer is that tumors can evolve and develop resistance to targeted therapies, much as many bacteria are now acquiring resistance to antibiotics, by changing driver mutations they are dependent upon. As a result, it is important to have an arsenal of targeted therapies with distinct mechanisms. In combination with new developments like DNA sequencing of patient tumor samples to inform treatment, this assortment of targeted therapies will enable oncologists to stay one step ahead of tumor resistance and meet the needs of a diverse human population.
One key characteristic that targeted therapies appear to share is a property called selective toxicity, meaning they are toxic to some cell lines of a given type but not all. The pathways used by cancer to grow are diverse, and different lineages have devised different strategies to overcome the restrictions on cell division placed on normal cells. As a result, what differentiates a particular lung cancer cell line from healthy lung cells is also likely to differentiate it from cancer cells of a distinct lineage. Because of this, molecules that exhibit selective toxicity are now being sought as starting points for developing new targeted cancer therapies.
Non-small cell lung cancer (NSCLC) accounts for about 85% of lung cancers and tend not to respond well to traditional radiation and chemotherapy. Furthermore, targeted therapies are currently only available to a subset of NSCLC patients – those with driver mutations for which such therapies have been developed. As a result, a significant need exists for new drugs targeting different drivers in NSCLC. Having previously used chemical strategies to identify the targets of several selective NSCLC toxins, Nikhil Madhusudhan and coauthors at UT Southwestern in Dallas used a photocrosslinking study to identify the targets of the compound SW069087, a quinazoline dione compound (abbreviated QDC in the original paper) that had been previously identified in a screen for selective lung cancer toxins (McMillan et al., Cell, 2018) (Fig. 2).
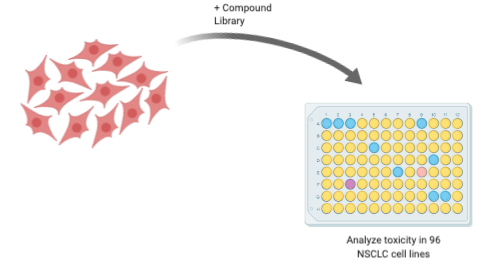
Figure 2: Compounds are tested for toxicity in 96 different NSCLC lines to determine whether they exhibit selective toxicity. –Justin Donnelly
A photocrosslinking study is a strategy used to identify the biological molecules a bioactive compound interacts with inside a cell. It relies on a photochemical probe, an analogue of the compound of interest that also includes a chemical motif that, upon exposure to ultraviolet (UV) light, can form covalent bonds with nearby proteins. The UV light excites electrons in a photo-reactive chemical group, in this case a benzophenone, causing cleavage of one of the C-O bonds and forming a reactive diradical, which can form bonds with proteins by replacing hydrogens bonded to carbon and nitrogen atoms. The third component in the photochemical probe is an analytical handle, in this case the alkyne (C-C triple bond). Alkynes are used to perform biorthogonal “click chemistry” with chemical groups called azides, which is a chemical reaction that can be performed in the presence of biomolecules without disrupting or reacting with them (Fig. 3). Depending on the experimental application, azides attached to fluorescent dyes or handles for separating the probe’s targets from the rest of the cell’s components may be “clicked” onto the alkyne.
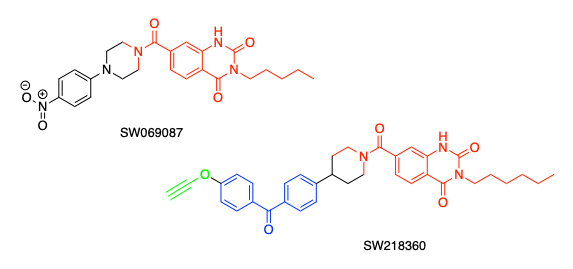
Figure 3: The hit compound from the NSCLC selective toxicity screen, SW069087, contains a quinazoline dione group (red). The authors produced a photoreactive analogue by elaborating the hit compound with a benzophenone photocrosslinking group (blue) and an alkyne (green) for attaching analytical molecules like fluorophores or biotin. –Justin Donnelly
Because the chemical group biotin binds very strongly to the bacterial protein streptavidin, this interaction is commonly used to enrich proteins from biological mixtures for subsequent analysis. Therefore, after initiating the photocrosslinking reaction, the authors lysed the cells, used the click reaction to attach biotin, and then analyzed the proteins that were enriched by their probe via mass spectrometry, which can identify molecules based on their molecular mass. As a control, the authors treated another group of cells by treating with both their probe and the original hit compound, with the rationale that if the hit compound out-competes the probe for a protein, it is likely to be a target of that hit compound (Fig. 4). They identified two proteins from their experiment that were strongly enriched in the probe-treated sample but were still able to be competed by the original hit compound. These were MT-ND1, a protein in the mitochondrial complex I of the electron transport chain (ETC), and HADHA, an enzyme involved in the consumption of fatty acids to release energy.
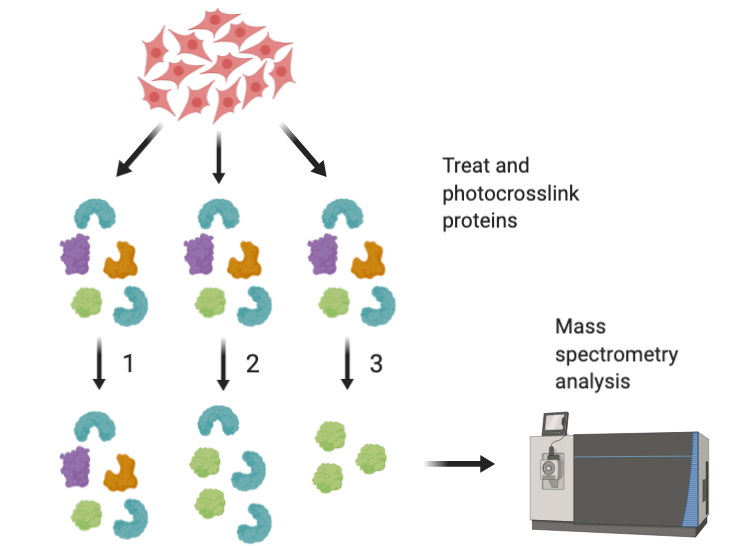
Figure 4: Cells are lysed and treated with either 1. No photocrosslinking probe, 2. Photocrosslinking probe only, or 3. Photocrosslinking probe and original hit compound (SW069087) and then analyzed by mass spectrometry. Proteins that are enriched in lane 2 and lost in lane 3 are likely to be targets of the hit compound. –Justin Donnelly
The ETC is a series of large protein complexes found in the inner membrane of mitochondria, often called the “powerhouse” of the cell. While originally recognized for their role in energy generation, mitochondria have more recently been appreciated to have broad functions, including an extremely important role in cell death pathways. The ETC oxidizes different carbon sources from metabolism – in other words, it removes electrons from them – and transports those electrons through a series of carrier complexes in the inner membrane to their final acceptor, molecular oxygen (O2), which produces water. In each step, the energy of these electrons is slightly reduced, and the complexes in the ETC can harness this energy to eventually drive the synthesis of ATP, the cell’s main energy currency. Interestingly, complex I is not an RTK and is not currently a target of cancer therapy. As a result, if a targeted therapy were to be developed it could have major impact on patients who may not respond to currently available targeted therapies like RTKis.
Just because a molecule is capable of binding to a protein does not imply that this binding event is responsible for a particular biological effect like toxicity. Therefore, it was also important for the authors to confirm whether either of these enriched proteins were actually involved in the selective toxicity observed during the screen. To accomplish this, they used CRISPR-Cas9 technology to silence the expression of HADHA and showed that the toxicity of the original hit compound SW069087 was not affected. This indicates that HADHA binding is not responsible for the molecule’s toxicity. MT-ND1 cannot be knocked out due to the importance of complex I for cell survival, so the authors used the MT-ND1 inhibitors pericidin and rotenone to block its activity instead. These compounds reduced the toxicity of SW069087 and also competed with the photocrosslinking analogue for MT-ND1 binding. As additional confirmation, the authors compared oxygen consumption (an indicator of ETC activity) in different NSCLC cell lines and found that cells displayed nearly identical oxygen consumption profiles as those treated with other complex I inhibitors. Taken together, these data indicate that the hit compound’s interaction with MT-ND1 inhibits complex I and the ETC and is responsible for its selective NSCLC toxicity.
This work clearly adds credence to the hypothesis that compounds targeting complex I have the potential to serve as a new class of targeted therapies in NSCLC, and perhaps other cancers as well. Currently, several candidates targeting complex I are in clinical trials, but the authors here have also validated a new class of molecules which can interact with a member of this complex. This provides drug developers with an additional set of potential “lead compounds”, compounds they could use as starting points to develop a drug, should the current candidates fail. More data are needed on the specific tumor properties that dictate reliance on complex I, enabling identification of patients who would respond to complex I inhibition in advance of treatment. Future work will also examine whether these molecules are safe and effective as anticancer drugs in animals, crucial experiments before any molecule will be permitted to enter a human being. This paper lays the groundwork for these experiments that will hopefully lead to a new set of chemotherapy alternatives.
Other References:
- McMillan, E. A., Ryu, M. J., Diep, C. H., Mendiratta, S., Clemenceau, J. R., Vaden, R. M., Kim, J. H., Motoyaji, T., Covington, K. R., Peyton, M., Huffman, K., Wu, X., Girard, L., Sung, Y., Chen, P. H., Mallipeddi, P. L., Lee, J. Y., Hanson, J., Voruganti, S., … White, M. A. (2018). Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell, 173(4), 864-878.e29. https://doi.org/10.1016/j.cell.2018.03.028