

Article Title: Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds
Authors: André Fischer, Manuel Sellner, Santhosh Neranjan and Martin Smieško, and Markus A. Lill.
Journal: International Journal of Molecular Sciences
Year: 2020
Doi: 10.3390/ijms21103626
The coronavirus infection has taken over the world – it has paused normalcy, wreaked havoc, and placed insurmountable pressure on healthcare. The urgency to control the crisis has led to the creation of successful diagnostic tests and the recent development of vaccines, but without the cure, we only treat the symptoms of the infection.
To find a cure, it is important to first identify a target protein, enzyme or receptor within the virus which when tampered with, does not allow the virus to carry out functions necessary for its survival. Researchers can then explore the chemical space in search of a ligand (molecule) that binds to the target in the virus. However, it is impractical to experimentally prod through this vast chemical space. Instead, using virtual screening, computational tools can rapidly reduce the million compounds to a handful of promising leads.
The scientists at University of Basel identified and targeted the main protease enzyme – which breaks down polyproteins to essential proteins – in SARS-CoV2 virus. By hampering this breakdown, the virus cannot carry out basic functions including reproduction. Since successful HIV and Hepatitis C treatments involve inhibition of proteases, the same strategy was suggested for the SARS-CoV2 strain. To this end, the scientists virtually screened 606 million compounds using the crystal structure of the SARS-CoV2 protease enzyme-inhibitor that was solved in January 2020.
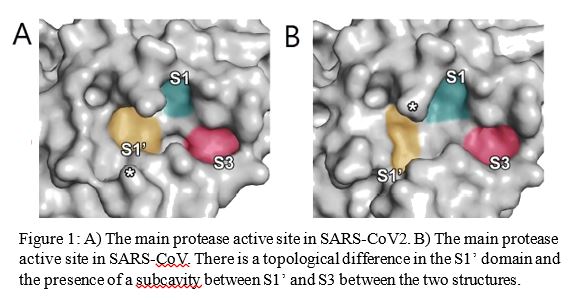
First, scientists tried to determine if compounds developed against SARS-CoV could be used against SARS-CoV2 by comparing their protease enzyme crystal structures. The enzymes in both the viruses showed a 96.1% sequence similarity, and a difference of one amino acid at the active site (the site on the enzyme which binds to the substrate to carry out essential reactions). Topologically, SARS-CoV2 has a subcavity (pocket) not present in SARS-CoV (Figure 1). Thus, compounds developed against SARS-CoV could only be used as a template.
The three-dimensional forms of the compounds were extracted from the ZINC database, a library of commercially available chemical compounds, and screened for shape similarity against known structures of SARS-CoV protease inhibitors. This reduced the dataset to 14,240 compounds, which were then docked against structures of the protease enzyme using the smina docking protocol. Smina docking is an automated process that ‘scores’ the interaction of the most stable ligand-enzyme complex by trying multiple binding positions. Based on the score threshold of -7kcal/mol, scientists selected 5,490 compounds that formed stable complexes and reacted favorably with the protease enzyme. Subsequently, Glide SP docking protocol which predicts the enzyme-ligand binding was used to increase confidence and cross-docking was done to validate the protocols and reduce the number of compounds. In the glide protocol, compounds with a score below -6.5kcal/mol were structurally similar so, more compounds were selected based on extended connectivity fingerprints (features representing molecular features like substructures, stereochemical information etc.) Finally, 2,783 compounds were obtained and clustered based on their molecular similarity calculated by a Tanimoto coefficient.
From each cluster, pharmacokinetic properties of the two best glide scoring compounds were calculated and based on the Lipinski and Veber criteria, 144 compounds were selected for final molecular dynamic (MD) simulations and free energy calculations. These criteria eliminates compounds with H-bond donors, acceptors, and coefficient of lipophilicity more than 5 and molecular weight above 500, as they are not ‘drug-like’.
MD simulations account for interaction properties like steric, ionic, covalent, vibrational etc. to calculate the thermodynamic binding free energy (ΔG). Using a MM/GBSA protocol, 29 compounds with predicted binding free energies better than the co-crystallized ligand were selected. 13 compounds were found to be toxic. 12 compounds were characterized for their oral absorption potential had excellent pharmacokinetic properties and were found to be efficacious. They interacted with the protease through at least 1 hydrogen bond, averaging to 3 H-bond interactions. 5 of them had comparatively low toxicity and two natural compounds were identified- Rhamnetin, whose preparations already exist in the market and (+)-Taxifolin, commonly found in plants.
All the compounds identified are only leads. Experimental studies to evaluate binding affinity, kinetics, and synthesis feasibility are still needed. But, by identifying promising leads the researchers have, at the time of publishing, given us a starting point in the hunt for a cure.
This is just one example of how computational tools can be used to solve important societal challenges. Computational tools in chemistry have been becoming more sophisticated and continue to address the challenges of molecular representation, property predictions, structural representation, and much more. Now, it has gained momentum due to the different ways of representation, advanced docking protocols, more data, and more powerful computational models. We know that by harnessing the power of computational speed, we can accelerate the drug discovery process and mitigate future crises.
Featured Image: Image by Miguel Á. Padriñán from Pixabay