
Title: Engineering Atypical Tetracycline Formation in Amycolatopsis sulphurea for the Production of Modified Chelocardin Antibiotics
Authors: Lukezic, T.; Fayad, A.A.; Bader, C.; Harmrolfs, K.; Bartuli, J.; Grob, S.; Lesnik, U.; Hennessen, F.; Herrmann, J.; Pikl, S.; Petkovic, H.; Muller, R.
Journal: ACS Chemical Biology
Year: 2019
URL: https://pubs-acs-org.libproxy.rpi.edu/doi/abs/10.1021/acschembio.8b01125
The world is at risk from antimicrobial resistant bacteria.
Only twelve years after Alexander Fleming discovered penicillin did the first antibiotic-resistant microbe, penicillin-R Staphylococcus aureus, appear. Since then, medicinal chemists have searched for alternative antibiotics to combat the growing resistance in common pathogens. Two professors who specialize in natural product discovery, Dr. Robert Muller and Dr. Hrvoje Petkovic, lead the collaboration that resulted in several tetracycline-derived antibiotics effective against Gram-negative and Gram-positive bacteria.
Dr. Rolf Muller, now at the Helmholtz Institute for Pharmaceutical Research Saarland, has worked with actimomycetes and myxobacteria to discover natural products and expand the antibiotic arsenal of the modern world for over twenty years. He previously collaborated with Dr. Hrvoje Petkovic, assistant professor at the University of Ljubljana and R&D director of Acies Bio, in 2016. Dr. Petkovic focuses on the biosynthesis of medically-relevant secondary metabolites for application to various cancers. In their paper, Lukezic et al. combined enzyme catalysis and traditional synthetic characterization to produce over twenty-one compounds derived from chelocardin, a natural product of the actinomycete Amycolatopsis sulphurea.
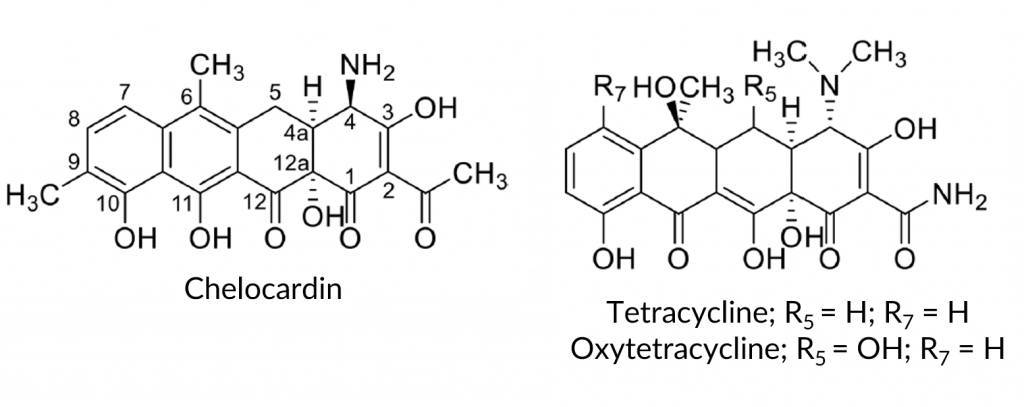
Chelocardin has broad-spectrum antibiotic activity and shares the same polyketide backbone as tetracycline, a common antibiotic for urinary tract infections and acne (Figure 1). However, there are several notable functional group differences between the structures: the acetyl moiety on the C2 carbon vs. a carboxamido, the primary amine with R designation on the C4 carbon vs. a tertiary amine with S designation, the lack of a hydroxyl group on the C6 carbon, the methyl group on the C9 carbon, and a different aromatization pattern, seen clearly at the C11 and C12 carbons (Figure 1). A. sulphurea produces chelocardin via an aromatic type II polyketide synthase (AIIPKS), which also produces tetracycline-backbone molecules such as oxytetracycline and chlorotetracycline. The authors hypothesized that by introducing a heterologous (non-native) system into A. sulphurea, the bacterium would produce other chelocardin derivatives through backbone modification.
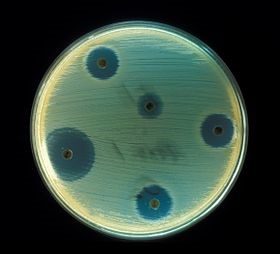
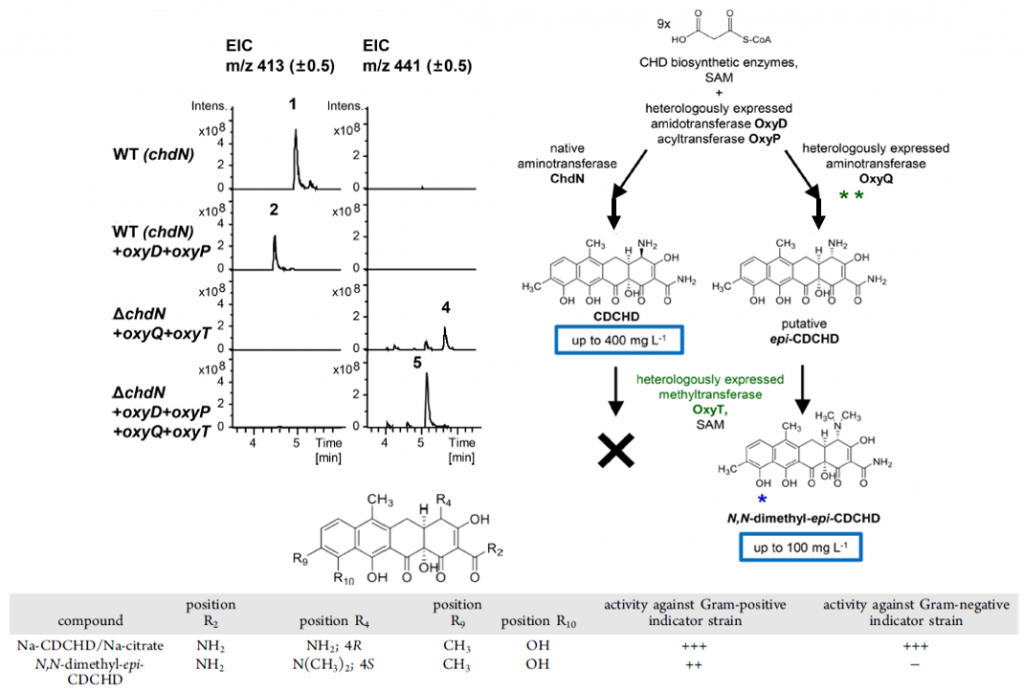
Lukezic et al. first showed the utility of this hypothesis in 2015, when their A. sulphurea mutant produced 2-carboxamido-2-deacetyl-chelocardin (CDCHD) in high yields (≤400 mg/L) and demonstrated significant antimicrobial activity against multidrug-resistant bacteria isolated from hospitals. To discover other tetracycline-backbone products, they fed chelocardin and CDCHD into an A. sulphurea mutant with the oxytetracycline (OTC) operon from Streptomyces rimosus; this gene cluster contains twenty-one genes (oxyABCDJKNPFELQTS just to name a few) and two flanking (genes before or after the gene cluster) resistance genes (otrAB).
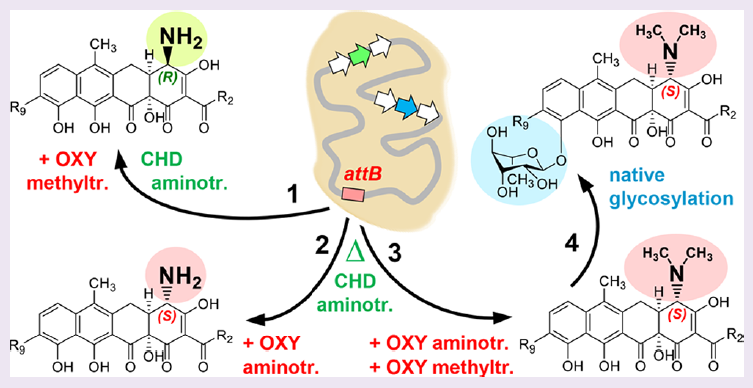
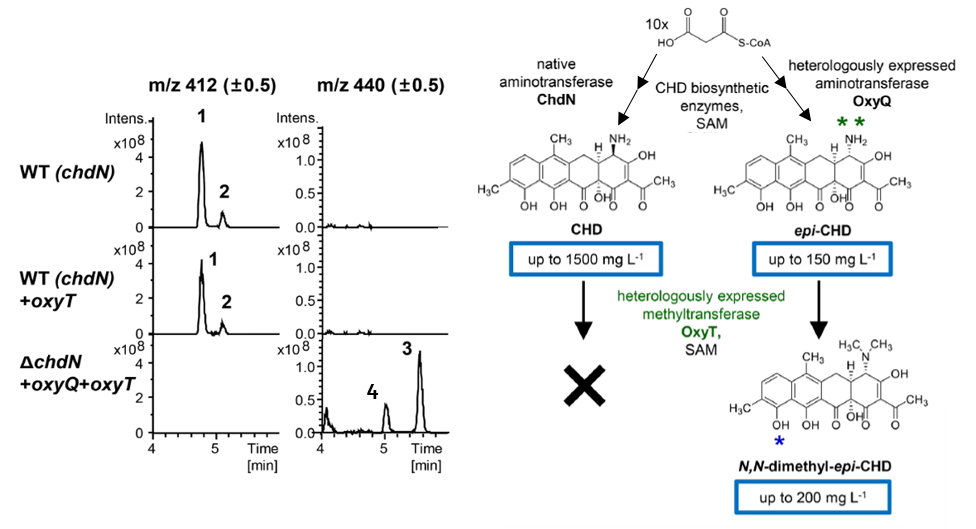
The authors attempted to introduce the S. rimosus oxyT gene, an S-configuration dimethyltransferase, into A. sulphurea – with no resultant chelocardin or dimethylated chelocardin. Based on the number of similarities between oxytetracycline and chelocardin (Figure 1), the authors speculated that the OxyT enzyme has narrow substrate specificity. Specifically, the stereo-configuration (i.e. the spatial arrangement) of the amino group must be S, like in oxytetracycline, and not R, like in chelocardin. Following that hypothesis, the authors inactivated A. sulphurea’s natural R-configuration C4-aminotransferase gene for chelocardin (chdN) and re-introduced oxyT and oxyQ, the OTC operon C4-dimethylaminotransferase. This produced the S-configuration, C4-dimethylamino product, but also the epimerized R-configuration product N,N-dimethyl-epi-chelocardin (Figure 2), identified via mass spectrometry and confirmed using various nuclear magnetic resonance methods. Epimers are a set of two compounds that differ only in the spatial arrangement aka stereo-configuration of one of their functional groups.
Introducing the amidotransferase-endocing oxyD and putative (i.e. not confirmed) acryltransferase/thioesterase-encoding oxyP to the ΔchdN/oxyQ/oxyT operon led to production of N,N-dimethyl-epi-2-carboxamido-2-deacetyl-chelocardin (CDCHD; Figure 3). The delta symbol (Δ) is a short-hand for how microbiologists name mutants; the genes following the Δ symbol are what genes the mutant lacks. Most interestingly, the epi-CDCHD had moderate activity against the Gram-positive representative Staphylococcus aureus and virtually none against the Gram-negative representative strain Pseudomonas aeruginosa, while the unmodified CDCHD was significantly effective against both bacterial types. This reveals more than a mild antibiotic alternative for Gram-positive bacteria; it shows how increased substitution of the amine group at the C4 position causes a loss of activity (i.e. NH2 on CDCHD vs. N(CH3)2 on N,N-dimethyl-epi-CDCHD; Figure 4).
The authors also observed an unexpected phenomenon as they characterized the compounds produced from A. sulphurea – each compound also resulted in higher mass ions – identified later as their glycosylated analogues. Glycosylation is the process of adding a sugar molecule to a product, in this case the sugar rhamnose. The glycosylated products of the dimethylated CHD and CDCHD reached yields comparable to their original compounds (40 mg/L to 100 mg/L) while the glycosylated analogues of CHD and CDCHD were present in minor amounts. Additionally, these glycosylated analogues were stereospecific – only R-configuration was observed in the glycosylated CHD and CDCHD analogues at the C4 position. In contrast, CHD and CDCHD start in the S-configuration.
Furthermore, to investigate the stereo-bioactivity of various derivatives of chelocardin and 2-carboxamido-2-deacetyl-chelocardin, the authors synthesized multiple non-bioengineered products using traditional synthetic means. The resultant products notably lacked antibiotic activity, except CDCHD-AHA, CDCHD-FuA, and CDCHD-pFBA, which have alternative R groups on the amido moiety (AHA = aminohexanoyl; Fu = furoyl; pFBA = p-fluorobenzoyl). Though the tetracycline alternatives were only used against three bacteria (K. pneumoniae, P. aeruginosa, S. aureus), Lukeziv et al. selected the most representative bacterial threats in today’s world.
The authors demonstrated well how an interdisciplinary approach, fusing bioengineering and medicinal chemistry, augments our ability to tackle the growing epidemic of antibiotic-resistant bacteria. In addition, the novel selective and sensitive antibiotics produced in this study will provide necessary alternatives to treat tetracycline-resistant strains of P. aeruginosa, K. pneumoniae, and S. aureus.