

Title: Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery
Authors: Francesco Gentile, Vibudh Agrawal, Michael Hsing, Anh-Tien Ton, Fuqiang Ban, Ulf Norinder, Martin E. Gleave, Artem Cherkasov
Journal: ACS Central Science
DOI: https://doi.org/10.1021/acscentsci.0c00229
Drug discovery is a time-consuming and expensive process; it can take anywhere between 5-15 years and a couple of billion dollars of investment on average to take a drug from ‘laboratory bench to bedside’. Each year only 40-50 new drugs are approved by the U.S. Food and Drug Administration (FDA). The whole process of drug discovery starts with finding a ‘lead molecule’ followed by optimization of molecular structure to achieve the highest efficacy. Then comes the clinical trial phases on animal and human where optimization is also achieved in terms of dose and administration method. Finally, the drug goes through the FDA approval process, and once approved, it can be commercialized.
The first step of this extensive process – ‘Finding the lead candidate’ is very difficult and it still relies a lot on serendipity. One of the famous examples of serendipitous discovery is ‘Penicillin’ in 1928 by Alexander Fleming. Although these surprising discoveries are known to happen from time to time, there is a growing need to make the initial part of drug discovery process more efficient to lower the cost and time burden.
So, how can we make the process of ‘finding the lead candidate’ more effective and time-saving? One of the ways lead candidates can be found is by screening the molecules virtually before performing the actual experiment in the laboratory. To that end, ‘molecular docking’ is used extensively to analyze three-dimensional orientation of potential drug candidates when attached to a diverse range of drug targets (e.g. proteins present within the body). Molecular docking is a method that can predict the orientation and binding affinity between a molecule and a protein structure virtually and assign the ‘best-fit’ orientation. Generally, a narrow subset of molecules is selected after running a million molecules against a drug target based on their favorable docking structure in terms of higher binding affinity. These virtual screenings of millions of molecules produce lots of data, which are underutilized (especially unfavorable docking results i.e. the ones that could not/loosely bind to a particular target) at this moment. Deep learning has the potential of utilizing this whole dataset to speed up the process of discovering new drugs in future.
The next logical question is – what is deep learning and how can it be used here? ‘Deep learning’ methods usually recognize a pattern within large datasets. This pattern is used to create a predictive tool which can be used to analyze datasets from very different sources. In this particular case, deep learning can be used to predict chemical structures of a lead candidate against a drug target by utilizing favorable and unfavorable docking results.
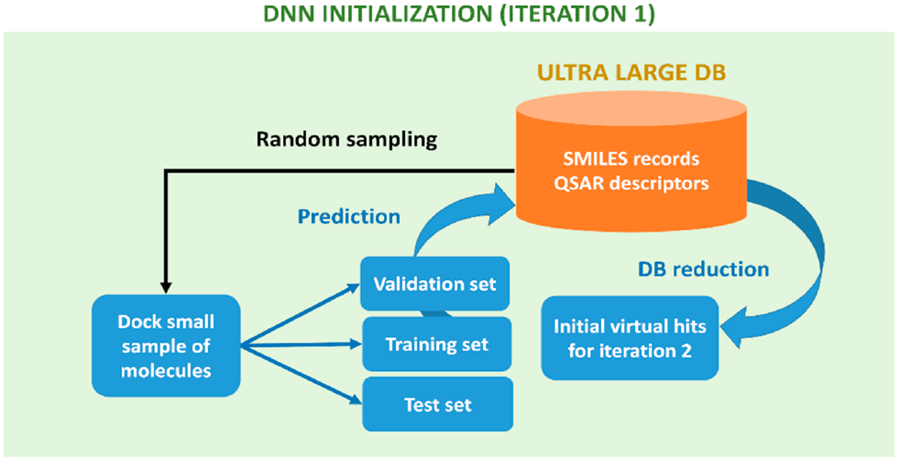
A team of researchers from the University of British Columbia, Canada introduced ‘Deep Docking’ (DD), which is a deep learning tool to virtually screen molecules rapidly and accurately (Figure 1). Initially, a set of quantitative structure-activity relationship (QSAR) descriptors (i.e. electronic properties of atoms/molecules such as electronegativity, dipole moment etc.; constitutional properties such as Molecular weight, Atom/bond counts etc.) is computed on a large database (DB). Then a small sample of molecules was randomly picked up from the database and the docking scores (favorable and unfavorable) of these molecules were linked to the corresponding structure-activity relationship descriptors through a deep-learning model. For example, let’s assume compound A showed favorable docking score (high binding affinity) and the same compound A has certain ‘structure-activity relationship’ properties in terms of molecular weight, number of atoms, electronegativity of compounds etc. Now, these ‘structure-activity relationship’ properties can be used as a metric to predict good docking behavior for other molecules in the database before performing the time-consuming ‘molecular docking’ itself.
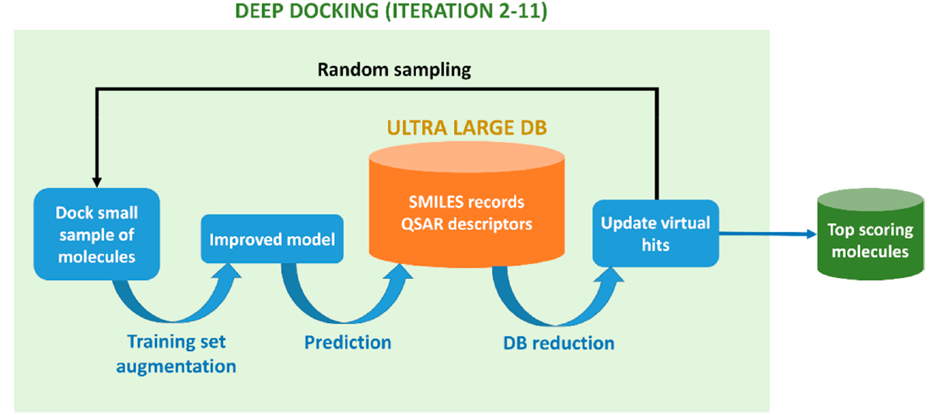
The resulting deep-learning model was utilized to predict docking outcomes of unprocessed entries in further iterations. In each iteration, the deep-learning model is fed with the outcome data of the samples and the model refines itself for further accurate prediction. This process stops after 11 iterations when the dataset is reduced significantly, and the top-scoring molecules were identified (Figure 2).
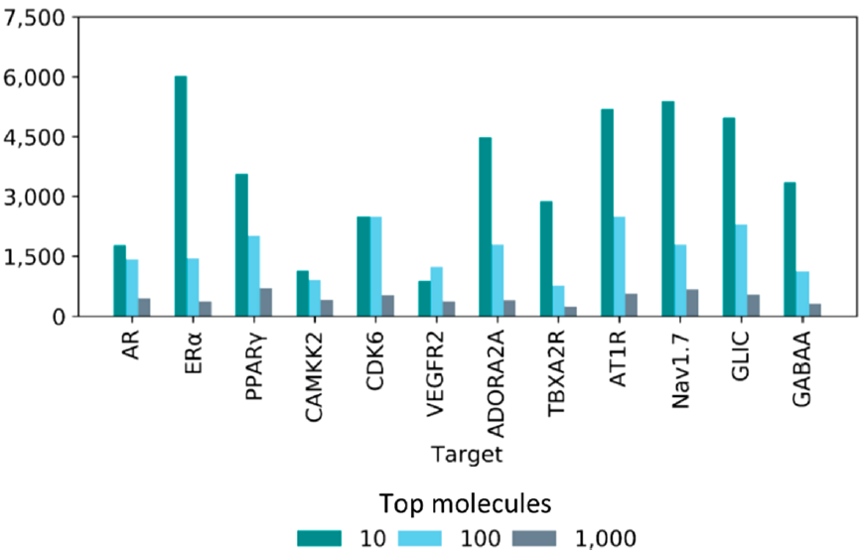
To identify the efficiency of the deep-learning model, an evaluation metric ‘enrichment factor’ (EF) is introduced. Enrichment factor of ‘top 10’ molecules against a drug target is defined as a ratio between the number of ‘True Positive’ results from those 10 molecules to the number of ‘True Positive’ results from randomly picked 10 molecules. ‘True Positive’ molecules are the ones that actually showed high binding affinity after molecular docking. Thus, higher enrichment factor corresponds to a better quality of prediction using the deep learning model. Figure 3 shows EF of top 10, 100 and 1000 molecules against different targets. The EF decreased consistently across all the targets suggesting that true hits are concentrated in the top-ranked molecules.
With increasing automation in the synthesis of drugs, more and more chemical structures are coming to the market, pushing ‘molecular docking’ to its limit in terms of time and cost. Using this deep-learning algorithm, new molecules can be found without running the ‘molecular docking’ on each of the molecules. Overall, this work provides a novel platform to be used by drug developers to expand the search for compounds for new therapeutics.
Cover Image: Gordon Johnson from Pixabay and Volodymyr Hryshchenko on Unsplash