
Title: Movement to the Clinic of Soluble Epoxide Hydrolase Inhibitor EC5026 as an Analgesic for Neuropathic Pain and for Use as a Nonaddictive Opioid Alternative
Authors: Bruce D. Hammock, Cindy B. McReynolds, Karen Wagner, Alan Buckpitt, Irene Cortes-Puch, Glenn Croston, Kin Sing Stephen Lee, Jun Yang, William K. Schmidt, and Sung Hee Hwang
Journal: ACS Journal of Medicinal Chemistry
Year: 2021
https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01886
Figures reproduced or adapted with permission through ACS Editor’s Choice. Further permissions related to this material should be directed to ACS. Featured image free to use by Pixabay license.
Imagine this…you’re a research scientist trying to develop a new drug. You want to inhibit a certain enzyme (usually a protein that binds a specific molecule to perform a chemical reaction) to treat a disease or decrease symptoms. You might have a petri dish of human cells containing the enzyme, or maybe you have the enzyme itself isolated for experiments. You test one molecule after the other and…hoorah! You found a molecule that stops the enzyme function! Is your molecule ready to be a drug? Not quite yet. In drug development, a functional molecule usually has to be altered before it is ready to be tested in humans.
In their recent research, Hammock and coworkers optimized a drug lead (promising potential drug) and even completed the first clinical trial for a molecule named EC5026 that they hope will eventually be approved to treat neuropathic pain. According to the Cleveland Clinic, neuropathic pain is a chronic condition where patients experience pain either spontaneously or from a usually non-painful stimulus (like cold or gentle pressure) due to damaged nerves [1]. About 30% of neuropathic pain cases are caused by nerve damage from diabetes, but other causes include Parkinson’s disease, multiple sclerosis, alcoholism, injury, and some chemotherapy drugs [1]. Current frontline drugs for neuropathic pain, such as pregabalin, are only somewhat effective and cause strong side effects such as drowsiness, dizziness, difficulty breathing, and decreased cognitive ability which frequently cause patients to stop treatment. Opioids—the addictive and dangerous class of painkillers—are often prescribed in addition to or instead of neuropathic pain-specific drugs, leading to potential opioid dependence. It is estimated that 7-10% of the general population suffers from neuropathic pain, so new drugs with fewer side effects are desperately needed.
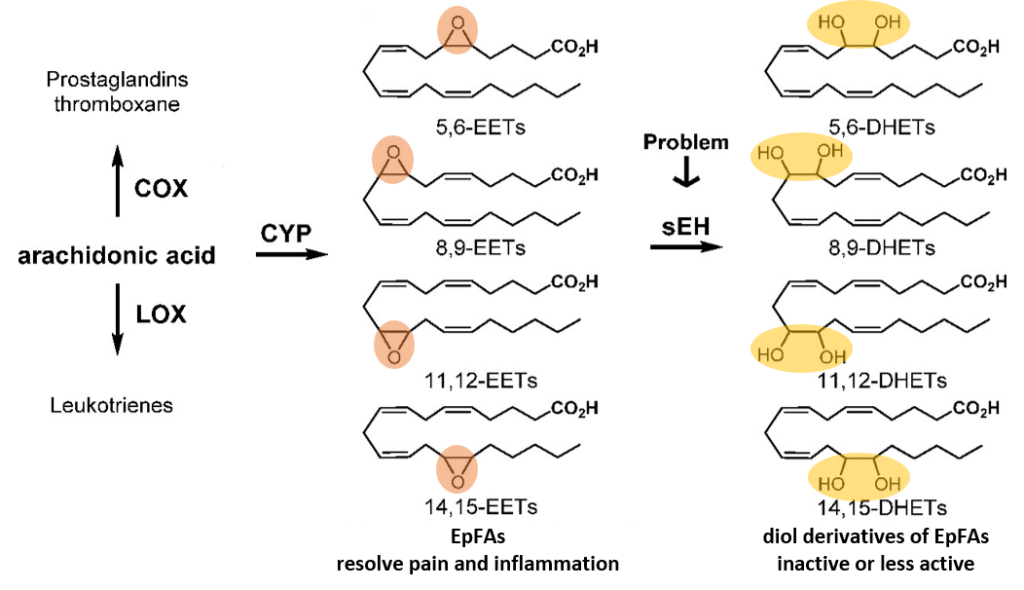
Many of the world’s drugs—including ibuprofen—act on a network of molecules in the body called the “arachidonic acid cascade” which consists of three branches. The first two branches (called COX and LOX) have been frequently targeted by drugs to block either the production or action of molecules that cause inflammation. However, the third branch—the cytochrome P450 (CYP) branch—was only recently understood, and thus can now be targeted by new drugs. In this branch, CYP enzymes convert arachidonic acid (and similar molecules) into Epoxides of polyunsaturated Fatty Acids (EpFAs) which decrease pain and inflammation (Fig. 1). However, enzymes called soluble Epoxide Hydrolases (sEHs) turn EpFAs into their inactive versions. Specifically, they break the epoxide (a 3-atom ring containing one oxygen and two carbons, Fig. 1, orange circles) into a diol (2 carbon atoms, each with its own hydroxyl or OH group, Fig. 1, yellow circles). Thus, for their drug development, Hammock and coworkers wanted to inhibit sEH enzymes to stop the degradation of the body’s natural anti-pain and anti-inflammatory molecules, the EpFAs.
From reading about previous studies trying to design sEH inhibitors, Hammock and coworkers knew that molecules that were too similar to EpFAs would be degraded very quickly by the body and wouldn’t remain in circulation long enough to be effective drugs. However, the molecule must still be similar enough to bind to sEH enzymes but without the epoxide needed for the sEH reaction. With this in mind, Hammock and coworkers developed a number of molecules where a central urea group (Fig. 2, blue circle) mimics the epoxide in EpFAs, and hydrophobic (water-repellent) groups would mimic the lipid nature of EpFAs (familiar sources of hydrophobic lipids are butter and oil). The scientists tested many similar molecules of this nature before deciding to move forward with EC5026 for clinical trials.
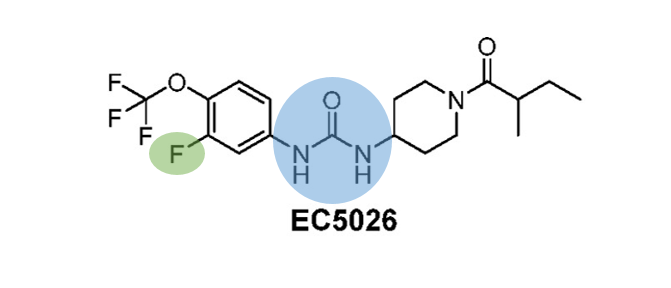
Many factors must be considered when assessing whether a molecule is fit to be a human drug including: potency (is the effect too strong or too weak?), bioavailability (does the molecule get into the bloodstream and then find its intended target in the body?), in vivo half-life (in a living organism, how long does the molecule remain in circulation before being degraded or expelled from the body?), and potential toxicity of degradation products. EC5026 was chosen for clinical trials in part because similar molecules tested had half-lives in animals that were too long for human trials. Among the candidate molecules, even one atom can make a huge difference in tuning a molecule for drug use. For example, the fluorine atom on EC5026 (Fig. 2, green circle) increased the potency of the inhibitor to an appropriate level. Tests in nerve-damaged rats showed that EC5026 was 20x as effective as the leading neuropathy drug, pregabalin. This fluorine also increased the potential bioavailability by decreasing the symmetry of the molecule and thus decreasing its tendency to clump together (clumps prevent individual molecules from inhibiting sEH enzymes). Because EC5026 doesn’t dissolve well in water alone, the drug was combined with polyethylene glycol (PEG) molecules— which increase solubility because they have water-attracting AND water-repelling regions and are commonly used in drug delivery—before being packaged in drug capsules for oral administration.
The first clinical trial of EC5026 in humans was recently completed. In this study, a small dose of the drug was given to healthy people to determine its safety. Once this dose was deemed safe, a higher dose was administered. Five different doses were tested, and happily, even at twelve-times the predicted necessary dose, no adverse side effects or toxic degradation products were seen. The road towards FDA approval is long and just beginning, but the results are promising. The next step for EC5026 is for each dose to be administered once daily for seven days to look for any side effects that might appear. Only then can the effectiveness of the drug begin to be tested on humans who have chronic pain conditions. Still, the progress of EC5026 is very exciting as a possible alternative to dangerous and less effective therapeutics.
[1] https://my.clevelandclinic.org/health/diseases/15833-neuropathic-pain