

Cover image: Pixaby, F1 digitals
Ryan Moreira, Jacob Wolfe and Scott D. Taylor, A high-yielding solid-phase total synthesis of daptomycin using a Fmoc SPPS stable kynurenine synthon, Organic and Biomolecular Chemistry 2021, 19, 3144 https://doi.org/10.1039/D0OB02504F
Some jargon and definitions:
- Antibiotic: A drug that is anti-bacterial, i.e. kills bacteria
- Peptide: A molecule that is a string amino-acids connected by amide bonds
- Depsipeptide: A peptide that has at least one ester linkage
- Lipo-depsipeptide: A depsipeptide that has a fatty-acid (e.g. a long (acyl) hydrocarbon chain) appended to it; lipo = lipid
- Protecting groups: These are chemical groups that are attached to a reactive functional group such as an amine, carboxylic acid or alcohol, that consequently reduces or eliminates the reactivity. These may include a Boc group, allyl ester or tert-butyl ether, respectively, but there are many others!
- Orthogonal protecting groups: Orthogonal protecting groups are those which can be selectively removed in the presence of another protecting group and therefore allow a chemist to unmask one reactive group at a time.
There’s a pretty high likelihood that you have consumed or at the very least, heard of, antibiotics. These wonder drugs protect us against bacterial infections which can come in many shapes and forms. Routinely, following a surgery a patient will be prescribed antibiotics as a precaution to ensure they do not contract a bacterial infection as their insides have been exposed to the outside world. You might also contract some devious bacterial freeloaders from everyday activities; perhaps you left the chicken on the bench for too long, your drinking water is contaminated, or you got a cut and didn’t keep it as clean as you could have.
Concurrently with globalisation and the rise of routine surgeries, bacterial infections have become quite common. Since the discovery of penicillin in the 1920’s, bacterial infections* have been considered more-or-less curable – that is, a routine hiccup that medicine can readily overcome. We now have an arsenal of antibiotics to target different types of infections. However, bacteria are clever – and with every new antibiotic we put to market, the bacteria evolve to resist our new drugs. This means that there are now bacterial strains which do not respond to any of our antibiotics and therefore, if you acquire an antibiotic-resistant infection, there is increasingly little a doctor can do to help you combat the illness. In some cases, this can lead to very dire and even fatal outcomes like those encountered in the pre-antibiotic era. This is both daunting and challenging for health professionals and medicinal chemists alike.
*Not all bacterial infections are bad, we live in harmony with many bacteria everyday.
Two of the largest challenges associated with developing new antibiotics is the ability to discover new types of antibiotics (read: interesting molecular structures), and the need to investigate the relationship between the chemical structure and medicinal activity (a structure-activity relationship, SAR, study). Naturally-occurring peptides are a particularly interesting source of novel antibiotics, including daptomycin (Figure 1) which has been shown to combat resistant strains of the gram positive bacteria, MRSA and VRE. Daptomycin (sold as Cubicin) is one of (the very few) new antibiotics put to market and represents a new class of antibiotic – the lipo-depsipeptides.
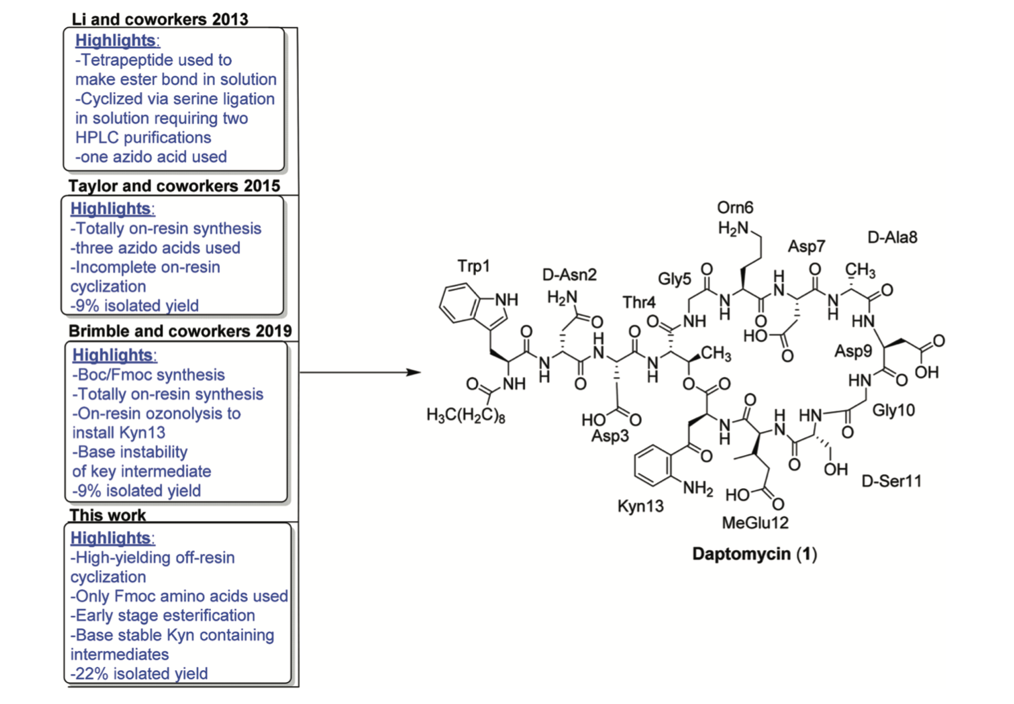
Daptomycin is currently produced biosynthetically – this means biochemists have (somewhat ironically) engineered a strain of bacteria to produce daptomycin to target its other bacterial brethren. Daptomycin is a particular challenging molecule to synthesise chemically. Peptides have historically been arduous molecules to prepare due to the large number of reactive groups present (see the chemical structure in Figure 1!). The advent of solid-phase peptide synthesis, which anchors the C-terminus of the peptide to a solid-support (e.g. polystyrene beads) and consequently eliminates the need for an additional orthogonal protecting group, has made peptides much easier to produce in bulk (Figure 2). In particular, the greatest benefit solid-phase synthesis provides over solution phase synthesis is the mitigating the need for a purification column following the addition of each new component through amide-bond coupling. Utilising a solid-support means that excess reagents can simply be washed off the heterogenous mixture.
Daptomycin (a lipo-depsipeptide) however presents a number of additional challenges which centre around forming the macrocyclic core. These include formation of an ester bond and installation of the unusual amino-acid kynurenine at this depsipeptide junction, and late-stage incorporation of the fatty acid tails. These hurdles have been overcome piece by piece, each with their own benefits and limitations, as we move toward a total solid-phase synthesis of daptomycin using only commercial reagents. See the further reading at the end of this article for more information on these developments. The most recent development in this crusade was by the Taylor group from the University of Waterloo in Ontario, Canada, who have executed an entirely Fmoc-based solid-phase synthesis of daptomycin. Fmoc solid-phase peptide synthesis is the most widely adopted form of peptide synthesis. The ability to prepare daptomycin using only Fmoc-protected building blocks, and not azido-masked amino-acids (as in the previous syntheses) which have to be synthesised in house, would streamline the preparation of daptomycin and allow ready access to more analogues.
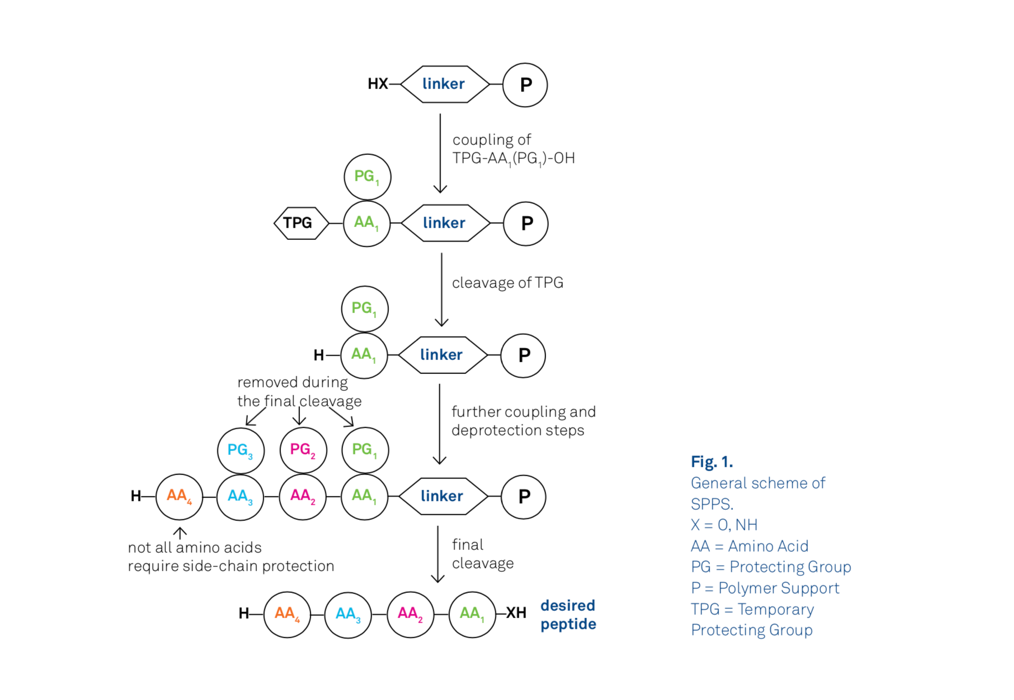
The key obstacle that was overcome in this study is the susceptibility of the kynurenine residue to undergo side-reactions in the presence of base (Figure 1). In Fmoc-based solid-phase synthesis, the C-terminal of the amino-acid is anchored to a polystyrene resin by an acid-cleavable bond, and the N-terminal amino-acid amine is protected by an Fmoc-group which can be removed to unmask the amine for subsequent reaction with the carboxylic acid of the next amino-acid. Fmoc-removal is carried out under basic conditions, generally with 20% piperidine in DMF. All other amino-acid side-chain groups are generally protected with an acid labile group that is removed when the peptide is cleaved from the solid-support with an acid such as TFA.
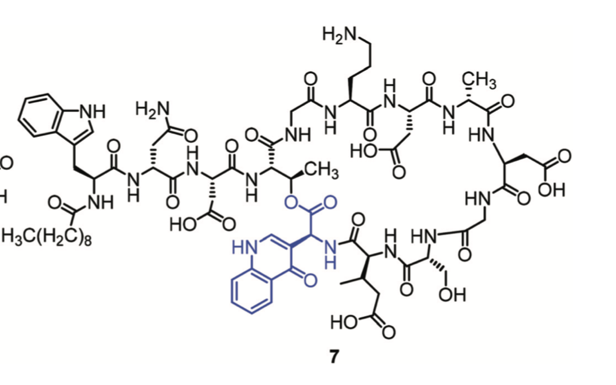
The aniline group of the unusual kynurenine is generally protected with two formyl (CHO) groups, or a tert-butoxy carbonyl (Boc) group and a formyl group. Though other groups have noted that the formyl protected kynurenine leads to side-products following multiple Fmoc-deprotections, the Taylor group set out to characterise these side-products. They observed side product was 10 mass units larger than the expected product and they hypothesised that the side-product could be an isocyanide derivative; however, they could not produce this analogue in a model system. A strong absorbance band at 330 nm lead them to investigate the idea that a quinolone moiety was forming (see the blue group in Figure 3). They postulated and then demonstrate that indeed a quinolone could form from the formyl protected kynurenine under these conditions, proceeding via a Camp’s cyclisation.
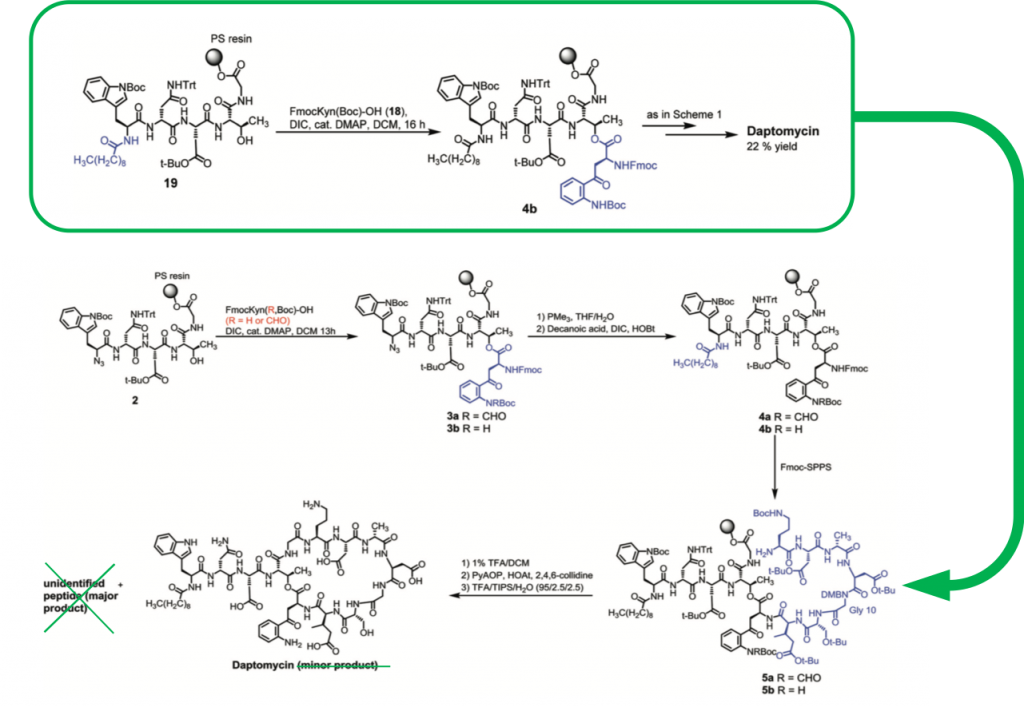
Importantly, they then went on to demonstrate that this side-reaction was highly dependent on the protecting group present on the aniline, and if only a Boc group was used to protect the aryl amine that the cyclisation did not proceed (see 4b in Figure 4, compared to 4a). This allowed the team to prepare daptomycin using only Fmoc-amino-acid building blocks in the highest yield currently reported for the chemical solid-phase total synthesis of daptomycin at 22% (Figure 1). Though this sounds modest, this is the final yield following HPLC purification of a molecule that requires approximately 30 reactions to furnish the macrocyclic 13 amino-acid lipodepsipeptide. This is an important step toward being able to prepare daptomycin in large quantities, and also enables new routes to access daptomycin and similar analogues, to ultimately, hopefully, develop new antibiotics based on this very useful peptide structure.
Additional reading:
- Lam, H. Y.; Zhang, Y.; Liu, H.; Xu, J.; Wong, C. T. T.; Xu, C.; Li, X. Total Synthesis of Daptomycin by Cyclization via a Chemoselective Serine Ligation. J. Am. Chem. Soc. 2013, 135 (16), 6272–6279. https://doi.org/10.1021/ja4012468.
- Lohani, C. R.; Taylor, R.; Palmer, M.; Taylor, S. D. Solid-Phase Total Synthesis of Daptomycin and Analogs. Org. Lett. 2015, 17 (3), 748–751. https://doi.org/10.1021/acs.orglett.5b00043.
- Xu, B.; Hermant, Y.; Yang, S.-H.; Harris, P. W. R.; Brimble, M. A. A Versatile Boc Solid Phase Synthesis of Daptomycin and Analogues Using Site Specific, On-Resin Ozonolysis to Install the Kynurenine Residue. Chemistry – A European Journal 2019, 25 (62), 14101–14107. https://doi.org/10.1002/chem.201903725.