

Article Title: A Non-Perturbative Molecular Grafting Strategy for Stable and Potent Therapeutic Peptide Ligands
Authors: Kathleen M. Sicinski, Vittorio Montanari, Venkata S. Raman, Jamie R. Doyle, Benjamin N. Harwood, Yi Chi Song, Micaella P. Fagan, Maribel Rios, David R. Haines, Alan S. Kopin, Martin Beinborn, and Krishna Kumar
Year: 2021
Journal: ACS Central Science
DOI: https://doi.org/10.1021/acscentsci.0c01237
A few years back, my relative became part of the 422 million people[1] worldwide to be diagnosed as diabetic. As a consequence, his household imposed several diet restrictions, and the terms “insulin” and “blood sugar” became common. Diabetics are immune-compromised and at risk of heart attack, blindness, kidney failure, and more, all of which make diabetes among the top 10 leading causes of death.[1]
When healthy humans consume food, human peptides – glucagon-like peptides (GLP1 and GLP2) and glucose-dependent insulinotropic polypeptide (GIP) – bind to their appropriate receptors, increasing insulin production and thus, our energy. In diabetics, insulin production is reduced as the protease, DPP4, cleaves and inactivates GLP1 before it can interact with its receptor. Administration of insulin doesn’t always treat diabetes, so doctors often prescribe medicines which mimic GLP1 to increase insulin production.
Every peptide has two ends; The beginning is the negatively charged C-terminal and the end is the positively charged N-terminal. DPP4 cleaves off the two amino acids, histidine and alanine, on the N-terminal, rendering GLP1 unfit for its receptor. Medicines that mimic GLP1 are modified to sneak by the protease, but they have low potency. In other words, for an effect to be observed, the patient must ingest very high doses. Now, Sicinski et al. have devised a modified GLP1 which is resistant to DPP4 proteolytic cleavage and just as potent as the native (naturally produced) GLP1.
Based on the cryogenic electron microscopy structures of GLP1 and DPP4, the researchers noted that the DPP4 inhibitor interacts with two glutamate residues on DPP4 (Highlighted in Fig.1a). Studies showed that mutants of DPP4 where the glutamate (negatively charged) is replaced by alanine (non-polar) cannot recognize GLP1. Thus, it was hypothesized that the glutamates interact with the positively charged N-terminus of GLP1. The researchers decided to remove this positive charge in GLP1 so that DPP4 can no longer recognize it.
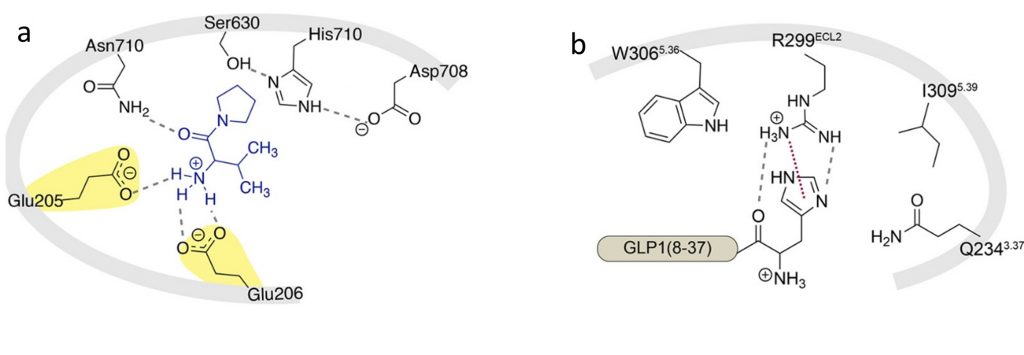
Acylation of the N-terminal formed a new acyl (amide) bond in GLP1 and increased its length (Fig.2). Although the protease DPP4 no longer recognized this GLP1 analog, the GLP1 receptor also failed to recognize it. Subsequent interaction studies between GLP1 and its receptor revealed that the histidine imidazole ring of GLP1 was important (Shown in Fig.1b), bulky groups at the N-terminal were tolerable, and the N-terminal primary amine of GLP1 was crucial to maintain potency. Based on this, they decided to alkylate the N-terminal. This way, the GLP1 analog retains the histidine side chain, the absence of a new amide bonds makes this analog tolerable, and the charge on the N-terminal is neutralized. The modified molecule was called 2-GLP1.
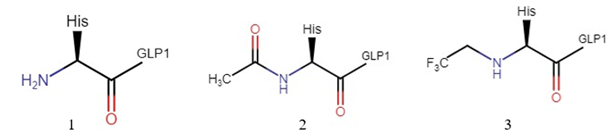
The potency of 2-GLP1 was remarkably close to that of the native GLP1. This was surprising because 2-GLP1 has a secondary amine, and it was previously thought that a primary amine was required for receptor binding. To check if DPP4 cleaved 2-GLP1, the researchers incubated 2-GLP1 and DPP4 and compared the LC-MS (liquid chromatography-mass spectroscopy) spectra of 2-GLP1 before and after. No change was observed, and thus the researchers concluded that 2-GLP1 was resistant to proteolytic cleavage by DPP4.
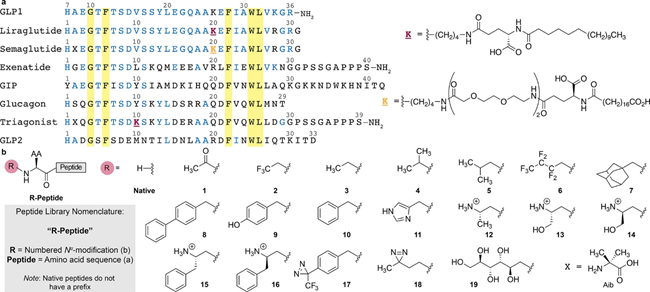
The same strategy was applied to other peptides: GIP (amplifies insulin production and modulates fat metabolism), GLP2 (ensures optimal uptake of intestinal nutrients), and glucagon (increases energy expenditure and weight loss when GLP1 and GIP receptors are stimulated). Though these peptides have dissimilar N-terminals, they are all cleaved by DPP4 when administered externally. Alkylation of these peptides made them resistant to proteolytic cleavage, and they all showed excellent potency. (2-GIP was as potent as native GIP, while 2-GLP2 and 2-glucagon were more potent than their native counterparts). The researchers also tried alkylating current clinical GLP1 analogs and a triagonist (one that stimulates all three GLP1, GLP2 and GIP receptors) and also found resistance to proteolytic cleavage and a high potency.
Besides this, they also explored the potency of other ethyl groups differing in hydrophobicity, polarizability, stereochemistry, electronegativity, and charge (modifications shown in Fig.3). This provided additional insight into the receptor binding pocket and an opportunity to further fine-tune the analogs. Studies in mice showed that biologically, the alkylated peptides behaved just like the native peptides. Efficacy studies demonstrated that 2-GLP1 was as efficacious as its native counterpart. In conclusion, 2-GLP1 is a desirable analogue of GLP1 working both in vitro and in vivo.
In this work, the researchers used alkylation chemistry and developed a novel method of conferring protease-resistance yet retaining receptor binding abilities. They successfully showed that 2-GLP1 can restore normal blood sugar levels and gained insight into the GLP1 receptor binding pocket. Furthermore, they showed that this methodology applies to other peptides in the same family. The hope is that this can help build a library of high-potency, protease resistant molecules. Though 2-GLP1 is not yet ready to enter the medicine market, it provides a lead to a better drug, and opens doors for more research on the application of this strategy to other receptor systems.
[1] Diabetes Fact Sheet, WHO. https://www.who.int/news-room/fact-sheets/detail/diabetes