

Title: Small molecule photocatalysis enables drug target identification via energy transfer
Authors: Aaron D. Trowbridge et al.
Journal: BioRxiv
Year: 2021 DOI: 10.1101
One of the most popular methods for screening for potential drug candidates is through phenotypic screens, in which the viability of drug candidates is determined by their morphological or biochemical effects on cell lines or model animals. For example, the death rate of a cancerous cell line could be used to find new anti-cancer drugs. This method is powerful because it is more unbiased and physiologically relevant than finding compounds that can bind to hypothesized drug targets in vitro.
However, once potential drug candidates are found, it is still important to find what their targets are to be able to improve the compounds and determine their mechanism of action; this process is known as target ID (Figure 1). Since there are hundreds of thousands of proteins present in the human proteome, this step can be extremely challenging and become a bottleneck in the drug development pipeline. A recent paper from the MacMillan group at Princeton introduces µMap target ID, a new technique that could significantly cut development time for new therapeutics by improving existing target ID methods.
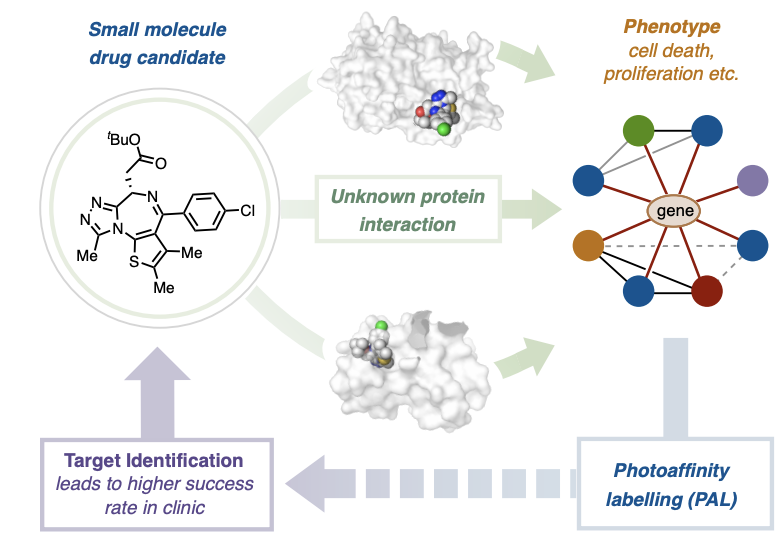
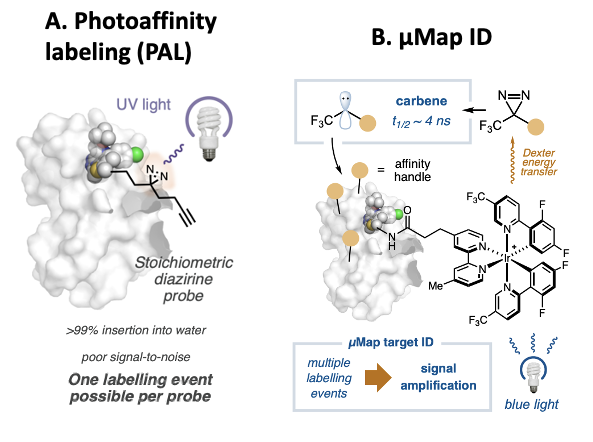
The standard method for target ID is known as photoaffinity labeling (PAL); it consists of modifying the drug candidate to include a click handle that covalently labels the protein the compound binds. More specifically, drug candidates are modified to include a handle with a diazirene and an alkyne, functional groups consisting of double bonded nitrogen and triple bonded carbon atoms, respectively. When a diazirene is irradiated with UV light, it dissociates from the compound, and the lone pair bound to a carbon atom (known as a carbene group) that is left behind can react with nearby molecules. In this way the drug can be covalently bound to the protein it is binding to.
The alkyne group can then be used to isolate the proteins that have been covalently tagged through a click reaction, as it can react and form a covalent bond with molecules with azide groups. Usually, the azide group is attached to biotin, which binds strongly to streptavidin and can be used to pull down the tagged proteins. Once the proteins have been isolated, they can be identified with mass spectrometry (figure 2a).
While PAL is a powerful technique, it suffers from some limitations. One large obstacle is that the carbene generated upon irradiation with UV light readily reacts with water. Therefore in >99% of the reactions, the drug candidate ends up bound with an alcohol group and unable to undergo another click reaction. Consequently, most of the drug present in a sample will not covalently bind the target protein, making the signal for the mass spectrometry step very low.
Meanwhile, with µMap target ID, the chemists replaced the diazirene/alkyne warhead with an iridium-based catalyst on the drug candidate, and made the warhead a separate molecule. The iridium catalyst transfers energy from irradiated blue light to nearby carbene atoms in a process that is known as Dexter Energy Transfer. Diazirene warheads that are far away from the catalyst are not affected by the blue light, so that carbenes will only be generated near the iridium catalyst. Thus, only the target of the drug-iridium compound will be covalently labeled by the carbene molecules (Figure 2b).
Just as in PAL, the alkyne handles can then be used to isolate the tagged proteins to be able to identify them. This technique’s main advantage over traditional PAL is that a single drug bound the protein can result in multiple labeling events, since the drug is not bound to the click handle but to the catalyst. Most carbenes still react with water, but since the iridium catalyst catalyzes the formation of many carbene molecules, the number of labeling events caused by a single drug bound to its target is much greater. This amplifies the signal of the labeled target which facilitates its identification through mass spectrometry.
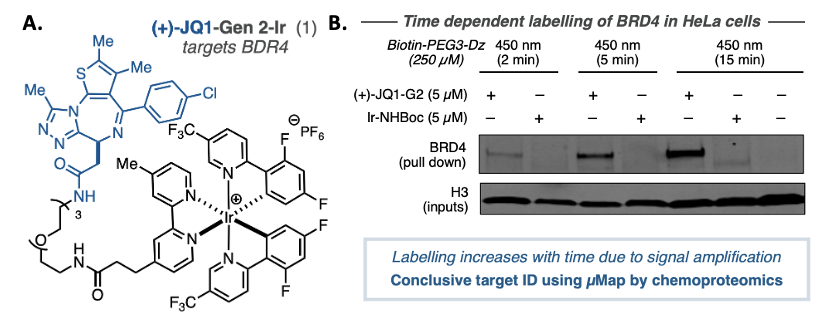
To validate the technique, the authors of this paper needed to use µMap ID with a drug whose targets are already well established, this allows them to compare their results to previous findings and confirm that the new technique is working as expected. They chose JQI, a molecule known to inhibit bromodomain proteins like BRD4 and is in clinical trials against several types of cancer. To use µMap ID, they linked JQI to an iridium catalyst, generating compound (+)-JQI (Figure 3a). It was important to verify that the catalyst did not interrupt the activity of (+)-JQI, so they measured its ability to bind bromodomain proteins post-modification and determined that the binding was virtually identical to the free JQI molecule.
Next, they moved on to measuring the activity of the compound in cellular assays. They incubated HeLA cells with (+)-JQI and an alkyne handle compound and found that it resulted in the labeling of the BRD4 protein via a western blot (Figure 3b). Incubating the cells with the iridium catalyst alone resulted in no labeling of the protein, proving that the labeling is due to JQI binding to its target protein. They used mass-spectrometry based techniques to identify all targets of JQI, and the targets that they found corresponded to the ones that are already established, thus confirming the accuracy of the technique.
µMap target ID has the potential to have significant impact in both academic and industrial labs due to its ease of use and ability to deconvolute the mechanism of action of potential therapeutic compounds. This paper shows how deep understanding of chemistry can lead to breakthroughs in biology and drug development.