
Title: Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration
Authors: Vighi, E., Trifunovic, D., Veiga-Crespo, P., Rentsch, A., Hoffmann, D., Sahaboglu, A., Strasser, T., Kulkarni, M., Bertolotti, E., van den Heuvel, A., Peters, T., Reijerkerk, A., Euler, T., Ueffing, M., Schwede, F., Genieser, H.-G., Gaillard, P., Marigo, V., Ekstrom, P., Paquet-Durand, F.
Year: 2018
Journal: Proceedings of the National Academy of Sciences
http://www.pnas.org/content/115/13/E2997
Inherited retinal degenerative diseases are a devastating class of disorders that cause the slow, inevitable decay of photoreceptor cells within the eye, eventually leading to permanent blindness. Unfortunately, the genetic diversity of this group of disorders has prevented the development of a targeted treatment platform. The location of the disease also renders many traditional drug delivery methods ineffective due to the presence of the blood-retinal barrier (BRB) in the eye. The authors of this paper sought to develop a new treatment system in response to these disease-specific challenges.
To understand how these scientists sought to combat retinal degeneration (RD), one must first learn a little bit about how the human eye works. Light is detected by two main types of photoreceptor cells in the eye, rods and cones. Rods are narrower and more sensitive to light (they can be triggered by a single photon!) but are less responsive to different wavelengths / colors of light. Cones require higher light levels but are more diverse in composition and respond to different light wavelengths, allowing humans to perceive color. Although humans have approximately 20 times as many rods as cones (120 million compared to a mere 6 million!), we rely primarily on cones since we operate in the brighter daylight hours.
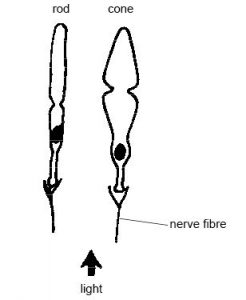
Many different types of RD tend to target rods first, depleting these cells before moving on to the more heavily-used cone cells. As a result, scientists opt to preserve these rod cells as a protective measure against further degradation; researchers have coined the adage “spare the rods, save the cones”[1].
With this in mind, the authors looked a little more closely at what might be causing RD on a molecular level. They hypothesized that a signaling molecule, cyclic guanosine-3’,5’-monophosphate (cGMP) is involved in downstream processes in the cell that are responsible for hereditary RD. cGMP interacts with two main effectors, cGMP-dependent kinase (PKG) and cyclic nucleotide gated ion channels (CNGCs), and is degraded by phosphodiesterase-6 (PDE6) in the cell. If PDE6 is mutated, cGMP rises to catastrophic levels and destroys all rod cells, with cone cells falling closely behind. The reason for this photoreceptor wipeout is due to overactivation of PKG and CNGCs by excess cGMP; one can think of it as a sewer blockage: if PDE6 is not funneling sewage (cGMP) out of the system quickly enough, normal decomposition processes (PKG and CNGCs) will be working overtime to generate a massive buildup in the system, causing the line to burst (cell death!).
To overcome this challenge, the authors designed a suite of cGMP analogues that act as structural mimics, binding and inactivating PKG and CNGCs to inhibit their negative downstream effects. The researchers tested monomeric and dimeric analogues of cGMP (some with previously reported activatory or inhibitory effects) in tissue culture, retinal explants, retinal slices, and (ultimately) in live rodents.
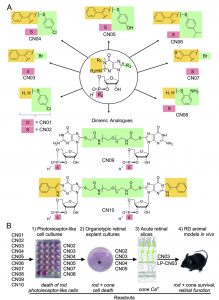
Initial tests revealed that six of the cGMP analogues (CN02 – CN07) prevented degradation in a cell line with PDE6 inactivated. These compounds produced statistically significant reduction in cell death at relatively low concentrations (50 uM) and did not diminish cell viability in wild-type tissue.
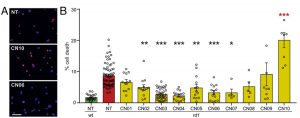
Further testing of the remaining analogues selected CN03 as a standout molecule due to the fact that it lowered calcium (Ca2+) levels in the cell, an important feature as high Ca2+ has also been correlated with photoreceptor degeneration after an extended period of time. The researchers verified that CN03 specifically targeted PKG and CNGCs and could also protect against RD in different cell lines with various hereditary mutations.
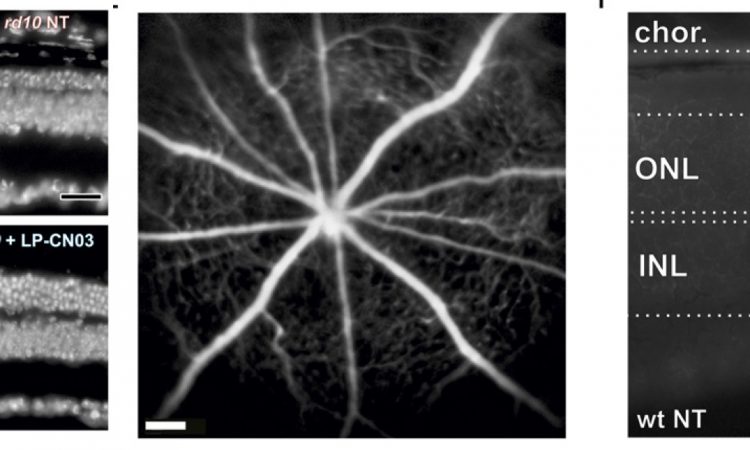
Thrilled with the initial success of CN03, the authors sought to design a delivery system to transport the drug across the BRB and into the retinal space. They chose to encapsulate the drug in a transport vessel (liposome) that could safely cross the BRB and degrade harmlessly within the interior of the eye.The liposome-packaged CN03 (LP-CN03) proved much more potent than a dosage of “naked” CN03, allowing increased efficacy and persistence of the drug within cells. The authors tracked photoreceptor cell numbers over time in live rodents and found that LP-CN03 produced a modest improvement over untreated mutant cell lines. They also conducted electroretinogram (ERG) tests of retinal activity and found that LP-CN03 offered significant improvement in the cone response when compared to an untreated sample.

In summary, this paper outlined a very clever new therapeutic system for treatment of RD in an animal model. The researchers’ initial results suggest that cGMP effectors are potential targets for new drugs to treat this destructive disease and may offer a solution to the heterogeneity of RD. Their liposome-based delivery system also shows promise as a method to unlock the ironclad BRB to other chemical-based therapeutics. We certainly hope that this paper brings us one step closer to providing an effective treatment system for retinal degeneration!
[1] Curcio CA, Owsley C, Jackson GR (2000) Spare the rods, save the cones in aging and age-related maculopathy. Invest Ophthalmol Vis Sci 41:2015–2018.