
AUTHOR: Song Hee Jeong, Hyun Ju Kim, Sang Jun Lee
JOURNAL: ACS Synthetic Biology
YEAR: 2023
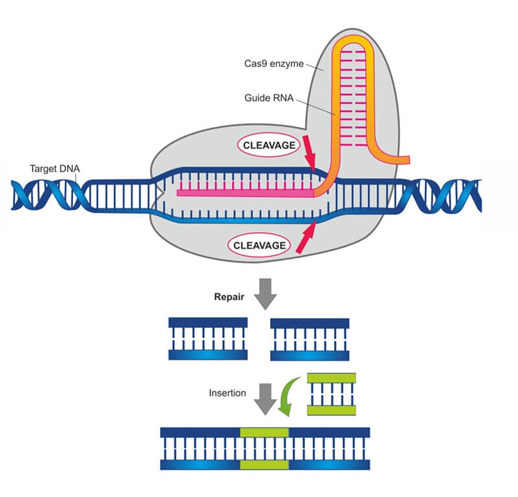
The bacterial immune system provided inspiration for the most popular genome editing tool – CRISPR-Cas9 (Clustered regularly interspaced short palindromic repeats-Cas9). Bacterial DNA has an anti-viral arsenal in the form of CRISPR arrays. These are short, palindromic repeats separated by viral DNA. When bacteria are infected with a virus, it produces RNA segments that recognize and attach to the viral DNA from this CRISPR array. A genetic scissor called Cas9, produced by the bacteria, chops off the viral DNA. We adopted this system to edit genes in other cells. We use a plasmid that contains a guide RNA (sgRNA) that recognizes and binds to the gene that we want to edit out from the cells and the Cas9 enzyme, which then chops off the target DNA. Then, the natural DNA repair system mends the ‘cut’ in the DNA (Fig 1).
But what if we didn’t want to remove the gene of interest but just control its expression? The CRISPR interference (CRISPRi) system uses a modified Cas9 where it has lost its ability to chop off the target DNA (dCas9). The sgRNA binds to the target gene and acts as an artificial repressor by not allowing the DNA segment to be converted into the RNA. If we use multiple sgRNAs, we could regulate the expression of multiple genes at a time and gain knowledge of genes and pathways that are altered in different phenotypes. This is typically called a CRISPR screen. But doing a CRISPR screen requires the synthesis of multiple sgRNA’s with each being a minimum of 20 nucleotides long, which is expensive. Jeong et al. have found the shortest target recognition sequence (TRS) length that the sgRNA can be of in order to effectively repress the target gene and drive down the cost.
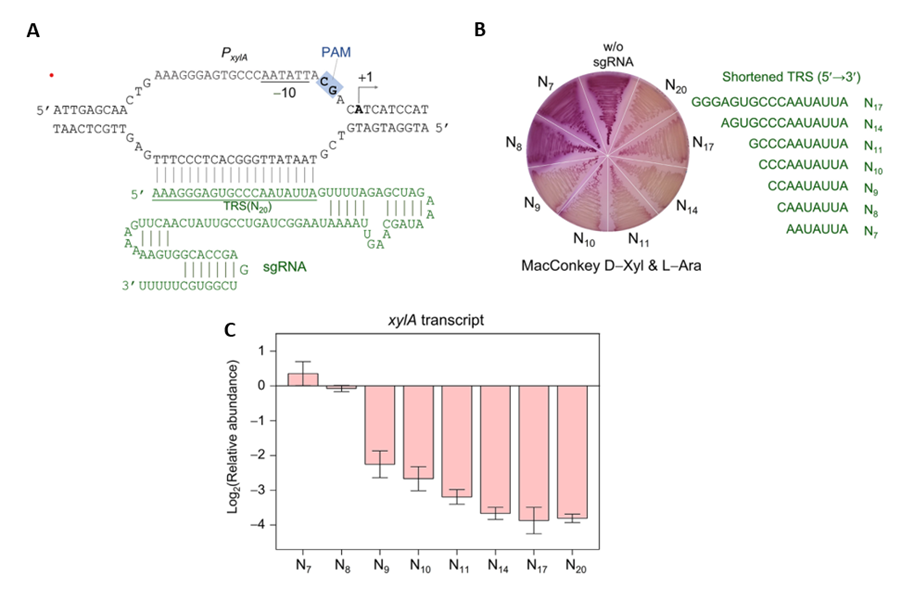
They targeted the xylA promoter gene, which transcribes the metabolic genes for D-xylose using different lengths of sgRNA, from 7 nucleotides to 20 nucleotides (N7, N8, N9, N10, N11, N14, N17 and N20). They generated these sgRNAs by removing nucleotides from the 5’ end of a 20 nucleotide sgRNA previously used (Fig. 2A). E. Coli was then transfected with a plasmid containing each of these sgRNA’s and the dCas9. The E. Coli was then grown into colonies in dishes. They found that plasmids containing sgRNA of lengths from 9 to 20 nucleotides produced white colonies, suggesting effective repression of the xylA promoter. Meanwhile, sgRNA of length 8 and 9 nucleotides produced red colonies, suggesting no repression (Fig. 2B). They confirmed that the xylA gene repression using Reverse Transcription Polymerase Chain Reaction (RT-PCR) (Fig. 2C)
They determined that the shortest length of the sgRNA required is 9 nucleotides.
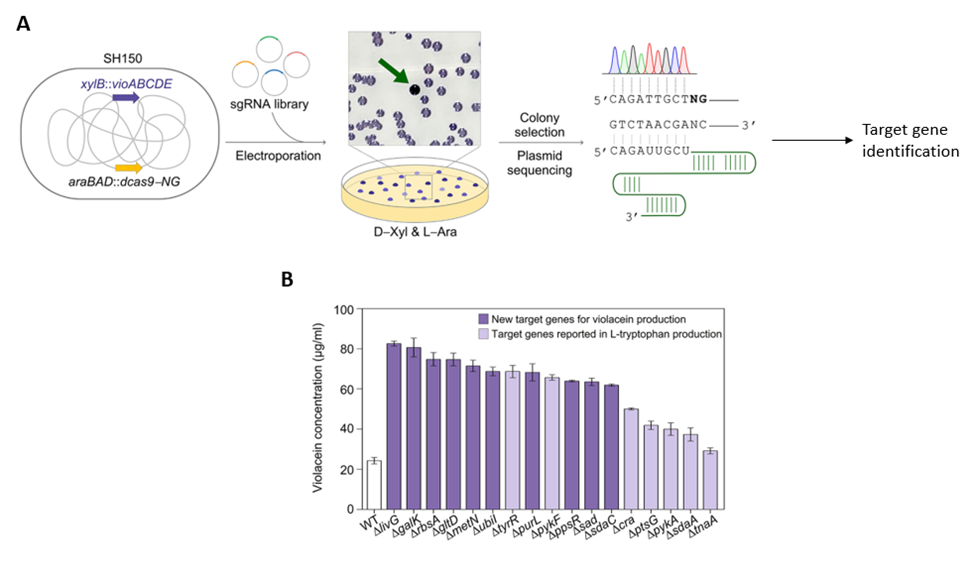
The earlier model looked at targeting a specific gene. To expand their model to identify genes related to a specific function, they looked at the production of violacein (the purple pigment that gram negative bacteria produce). In E. Coli, the gene vioABCDE produces violacein. So, they constructed a plasmid containing sgRNA’s of lengths 8 to 20, D-xylose inducible vioABCDE and the deactivated cas9, and they inserted this plasmid into an SH150 bacterial strain. They used a randomized, shortened sgRNA library. After allowing the bacteria to grow, they looked for the amount of purple pigment produced based on the RGB values. They identified that the N9 sgRNAs bound to 460 different sites while the others bound to only one target gene. Based on their criteria, they found 17 target genes, 5 of which were in the promoter region of vioABCDE, 7 genes were in the middle of the structural region and 5 genes were in the promoter and the structural region.
This research advances the evolution of the genome editing tool and paves the way for better CRISPR screens as it allows us to interrogate more genes using the fewer resources. Of course, more studies need to be done with a variety of targets and model cell lines to understand the limitations. I am also curious if we can use the shorted sgRNA for CRISPR-Cas9 genome editing. It has the potential to pave the way for the development of more high-throughput methods to study changes in the genome under different conditions thereby providing clues to different pathologies. Technologies like CRISPRi are constantly evolving to better our understanding of the genomic landscape, and there will no doubt be more advances to come.
References:
Jeong, Song Hee, Hyun Ju Kim, and Sang Jun Lee. “New Target Gene Screening Using Shortened and Random sgRNA Libraries in Microbial CRISPR Interference.” ACS Synthetic Biology 12.3 (2023): 800-808.