
Featured image and figure 2 from Chen et al., Science, 2018, 360 (6384), 71-75. Reprinted with permission from AAAS.
Title: Enzymatic Construction of Highly Strained Carbocycles
Authors: Kai Chen, Xiongyi Huang, S. B. Jennifer Kan, Ruijie K. Zhang, Frances H. Arnold.
Journal: Science
Year: 2018
http://science.sciencemag.org/content/360/6384/71
Organic chemistry enables us to push the limits of our curiosity to develop creative ways to build challenging molecular architectures. Sometimes, however, there are synthetic challenges that can only be addressed by biological macromolecules. The intricately precise spatial arrangement of reactive groups in a proteins active site, for instance, allows for regio-, chemo-, and stereoselectivity that very few isolated organic or metal catalyst can rival. This notion has spawned a field known as biocatalysis, which combines protein engineering with organic chemistry to generate variants of an enzyme that can perform a demanding organic transformation in high yields and selectivities.
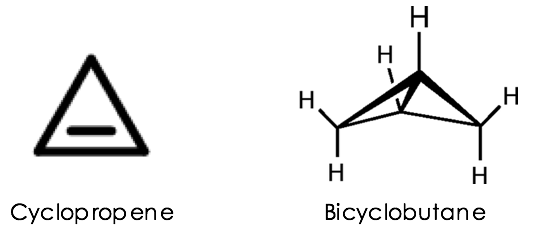
With biocatalysis becoming more mainstream in labs all across the globe, and with more taxing chemistry being both performed by and demanded from these engineered proteins, it has become hard to stand out in the field. Lately, however, the lab of Frances Arnold at Caltech elevated the biocatalysis standard by developing enzymes that can build some of the most strained tiny carbon rings (or carbocycles), cyclopropenes and bicyclobutanes (Figure 1). Despite their apparently simple structures, these carbocycles are the chemical equivalents of loaded springs, as they possess significant ring strain energies ranging from ~50-65 kcal/mol. The distortions in bond lengths and bond angles imposed by their geometry allow these molecules to undergo strain-release rearrangements to afford even more structurally rich scaffolds. Considering how unstable they are, it is laudable to see them being synthesized by traditional methods, let alone by engineered enzymes!
To attain their goal, the Arnold Lab decided to experiment with heme-containing proteins, which are known to have the substrate promiscuity and tunability needed for successful protein engineering. Through a process known as directed evolution, they generated E. coli bacteria that express regular or mutant copies of a heme protein. They then perform the chemical reaction of interest in the bacterial lysate and screen for product formation. Further mutations are performed on promising candidates, and the process is iterated until a very precise and robust enzyme is obtained. In brief, the group coaxed E. coli into creating incredibly challenging molecules by fine-tuning the bacterial genome, and thus, the protein that these genes encode.
After several rounds of screening and directed evolution of the promising proteins, they came upon a promising candidate in the form of a mutated variant of the protein cytochrome P411. This engineered enzyme, which they call E10, was able to generate an enantiomeric cyclopropene intermediate and then convert this into a bicyclobutane product via two subsequent processes known as carbene transfers (Figure 2).
Carbenes are very reactive carbon centers that have two bonds and two unshared electrons. This means that the carbene is neutral, but contains an unshared electron pair and an empty orbital at the same time, making it simultaneously nucleophilic and electrophilic (see molecule 2 in Figure 2). Thus, carbenes can be added to systems that can both accept and donate electron density, such as alkynes or alkenes to generate 3-membered rings. This is how Arnold’s engineered enzymes formed their products (Figure 2).
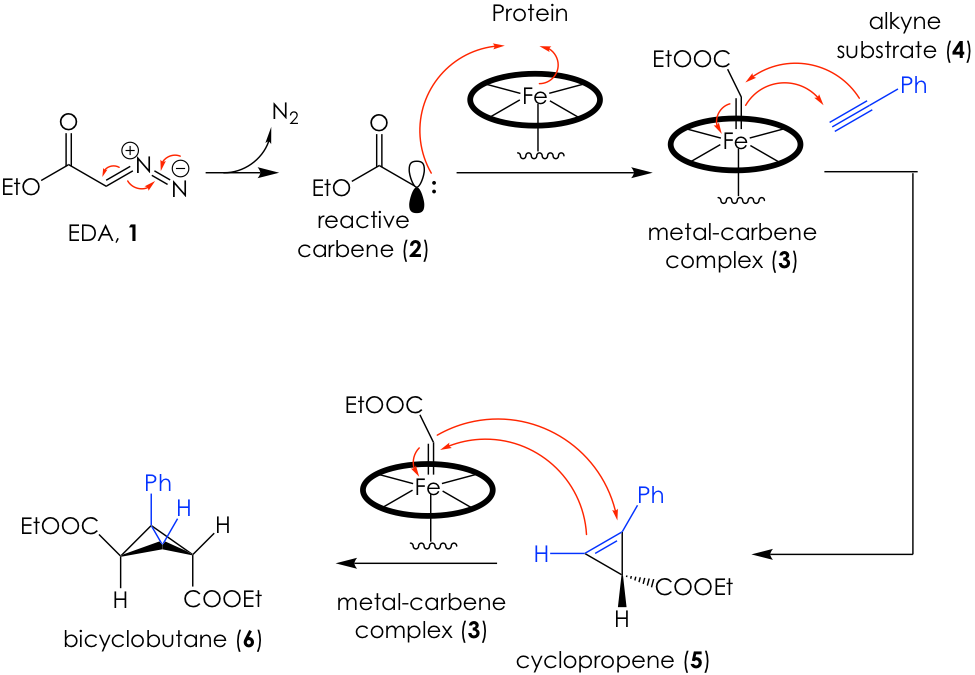
Briefly, they use a carbene surrogate known as ethyl diazoacetate (EDA, 1) to generate a carbene in situ (2), which can be trapped by the iron in the heme protein’s active site to create a metal-carbene complex (3). The carbene then adds across the pi system of an alkyne such as 4 to generate cyclopropene 5. The E10 enzyme is capable of stabilizing this unstable intermediate and subject it to another iron-mediated carbene transfer, this time across the double bond to produce the fused bicyclobutane product 6. Remarkably, all these reactions proceed enantioselectively. The curious reader is encouraged to refer to the original paper for a full recount of reaction design and screening.
Finally, the authors show that this reaction platform profits from a relatively generous substrate scope with respect to the alkyne. They also put to the test the reactivity of the bicyclobutane adducts by further derivatizing them, demonstrating that these strained cycles can tolerate esterification, click chemistry, and reductive conditions.
While it is not unforeseen that proteins can relatively seamlessly generate challenging bond formations seldom achieved by traditional means, it is remarkable to witness the development of enzymes that can forge very high-energy architectures not found in nature, such as the cyclopropenes and bicyclobutanes in this research. It will only be a matter of time to appreciate the next big step in biocatalysis.