
Title: Physical Chemistry of Epigenetics: Single-Molecule Investigations
Authors: Tae-Hee Lee
Journal: ACS Journal of Physical Chemistry B
Year: 2019
https://pubs.acs.org/doi/10.1021/acs.jpcb.9b06214
Featured image and figures reproduced or adapted with permission. Further permissions related to this material should be directed to ACS.
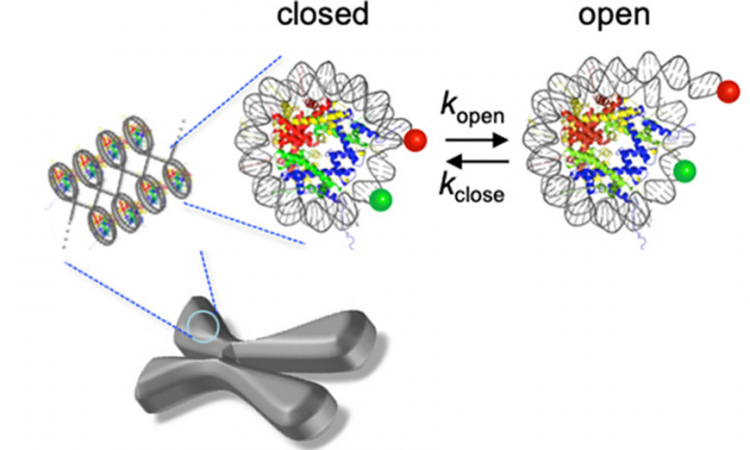
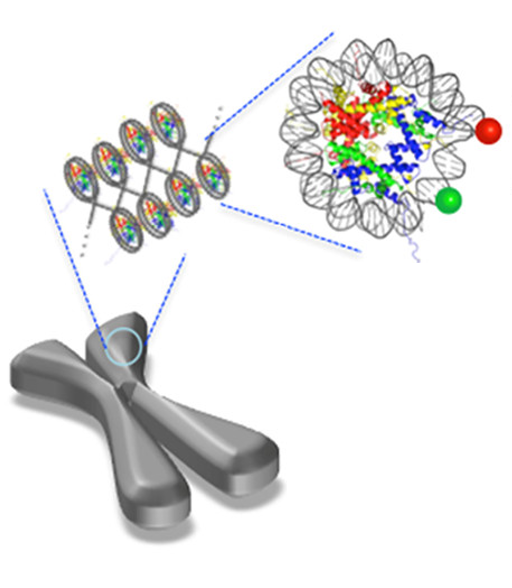
The human genome is huge—approximately 3 billion base pairs long—yet every one of your cells fits this entire genome into a space that’s about 0.7 femtoliters large (that’s 0.0000000000000007 liters!) In order to accomplish this extraordinary feat of packaging, the cell must utilize multiple levels of organization. First, the DNA is organized into small necklace-like beads by wrapping about 147 base pairs of DNA around a protein complex called a “histone core” (Fig1). Each DNA-wrapped histone core is called a “nucleosome core particle,” and this nucleosome is stable because DNA is a negatively charged macromolecule and is attracted to the positively charged parts of histones. The DNA is further organized by packing these nucleosomes closely together in a structure called chromatin, much like you might wind up a necklace or electrical cord for storage.
However, the genes in our DNA can’t remain tightly packed all the time; they need to be expressed through transcription and translation to make the proteins and enzymes that do the jobs needed to keep a cell alive. Therefore, the cell needs a way to control which genes are accessible to be expressed at different times. To accomplish this task, epigenetic modifications make up a layer of genetic control on top of (“epi” to) DNA’s genetic code. If your DNA is a cookbook that determines what recipes your cell could possibly make, epigenetic modifications are the head chefs that decide which recipes are on the menu for today. Despite the vital importance of epigenetics, scientists are still trying to figure out how these modifications work to regulate gene expression.
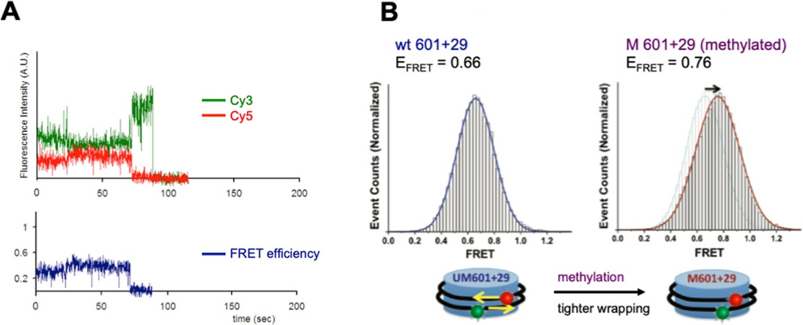
One of the most important epigenetic modifications is called CpG methylation. DNA is made up of four nucleotide bases (A, T, G, and C) linked together by a phosphate group (the “p” in CpG) and a deoxyribose sugar. An enzyme called a DNA methyltransferase can recognize when a C base is next to a G base, and this enzyme will covalently attach a methyl group (CH3) to the C base. These CpG methylations are known to decrease gene expression in the methylated region, but scientists are still investigating how they do this. Some studies have shown that other proteins recognize CpG methylations, and these proteins work together to tighten the overall packing of the chromatin. However, recently scientists wanted to test the hypothesis that the CpG methylations alone can somehow make the DNA wrap more tightly around a histone core particle. To do this, the scientists used single-molecule FRET (smFRET) techniques to look at a single nucleosome core particle with and without a CpG methylation.
FRET stands for Fluorescence Resonance Energy Transfer, and it requires two fluorescent molecules with specific absorbance and emission properties, for example, Cy3 which emits green light and Cy5 which emits red light. In FRET (Fig2A), a high-energy wavelength (for example, blue light) is absorbed by the first fluorescent molecule, Cy3. Cy3 emits a lower-energy wavelength (green). Cy5 can absorb this green light and emit red light, but only if Cy3 and Cy5 are close enough to each other in space. In the CpG experiment (Fig2B), Cy3 and Cy5 were attached to different locations on DNA wrapped around one histone core such that the two fluorescent molecules will move closer together (and give off higher FRET, i.e. more red light) if the DNA wraps more tightly around the histone. Just as the scientists had predicted, they saw more FRET when the DNA was methylated, indicating that a CpG methylation alone can physically cause tighter wrapping of DNA around a histone.
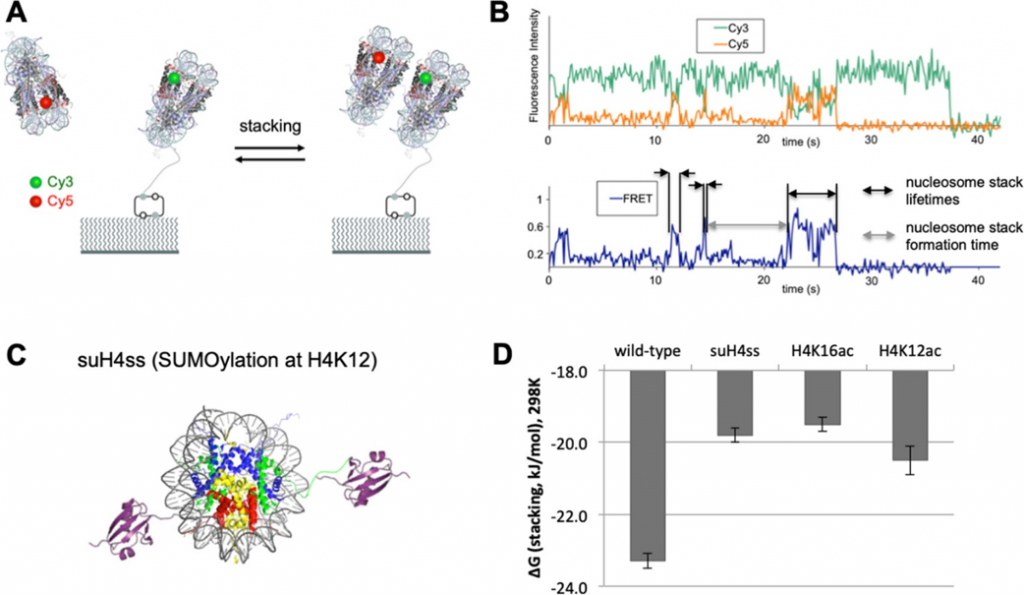
However, the scientists were also interested in how epigenetic modifications can affect the packing between two nucleosomes, since packing is necessary to form the overall chromatin structure. In this packing, one nucleosome will stack on another, so the scientists attached Cy3 to one nucleosome and Cy5 to another to determine how long it takes to form a “dinucleosome stack” (the formation time) and how long it takes for a dinucleosome stack to fall apart (the lifetime). This method was used to test the effects of two epigenetic modifications—acetylation and SUMOylation—where groups are covalently attached to the histone core particle instead of the DNA. In acetylation, a neutrally charged acetyl group replaces a positively charged group on the histone (specifically, a lysine amino acid) that usually stacks with a negatively charged group in another nucleosome’s histone. In SUMOylation, a very small protein called a SUMO is covalently attached to a histone’s lysine amino acid. This SUMO can recruit other proteins to further affect chromatin structure (much like a CpG does), but it is also very bulky, so scientists think SUMOylation alone can affect dinucleosome stacking. In their experiment (Fig3), the scientists tethered a Cy3-labeled nucleosome to a polymer surface, and let a Cy5-labeled nucleosome float around in solution, watching for the two to form a stack. Once again, their predictions were correct, and both acetylation and SUMOylation reduced dinucleosome stacking, likely due to changes in charge and shape (steric effects), respectively.
Many aspects of how epigenetic modifications work are still a mystery, but these experiments support the idea that they can change the accessibility of DNA for gene expression even in the absence of other DNA-organizing proteins. This work is very important, since increasing our understanding of epigenetics could be vital to understanding human development as well as tumor suppression. For example, CpG islands (DNA regions with a lot of CpG methylation) are known to suppress both tumor-related genes and organ development-related genes. Thus, the current research brings us one step closer to understanding the link between the simple genetic code and life as we know it.