

Cover image from Unsplash by Braydon Anderson.
Nicholas Barlow,* David K. Chalmers,* Billy J. Williams-Noonan, Philip E. Thompson,* and Raymond S. Norton*, ACS Chem. Biol. 2020, 15, 2070−2078
Chemists have become pretty good at designing drugs that bind a target, and affect some biological action. Initial testing of new drugs is largely carried out ‘in vitro’, which literally means “in a test tube”. In practice, this means a ‘simple’ lab test (not in cells or an animal), for example, testing how a drug interacts with its target, such as binding to a protein. However, this is just the first step in a long line of check boxes a molecule must tick, to graduate and become a fully-fledged drug. Clinical drugs need to be easily administered, absorbed, and navigate their way through the biological soup that is the human body, all while fending off the immune system and other biological process trying to break down the drug and eliminate it, to hopefully find their target and affect its therapeutic function. This is particularly difficult for large molecules, here Barlow et al. investigated a way of disguising large molecules so they can more effectively get into cells and reach their target.
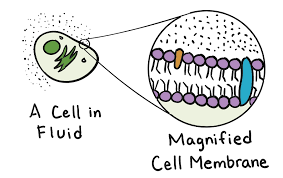
One of the last hurdles in this process is cell permeability. The large majority of disease targets require a drug to enter through the cell membrane to find their mark – however this is no mean feat. A drug must first be soluble enough in water that it can be immersed and transported through your 70% water-containing human body. The cell membrane, however, is made up of lipids – an oily outer bubble that defines the cell. So, an effective drug needs to be fat-soluble enough that it will happily meld with the fatty cell membrane, but not so fat-soluble that it doesn’t want to leave and can re-enter the water-filled cell on reaching the other side. Large drugs, such as peptides that target protein-protein interactions, do not conform to traditional permeability rules. Lipinski’s rule of 5 (Ro5), developed in 2001 by Christopher Lipinski working at Pfizer, was the gold standard for predicting the cell permeability of a small molecule drug. More specifically these rules are a predictor for whether a small molecule could be an orally-active drug. The Ro5 states that a molecule must have no more than five hydrogen-bond donors (HBDs, think alcohols and amines), no more than ten hydrogen-bond acceptors (HBAs, think carbonyls, and most nitrogen or oxygen containing functional groups), a molecular weight of less than 500 a.u. and a logP less than 5 (where logP predicts the lipophilicity or fat-solubility of a molecule and P is a partition coefficient).
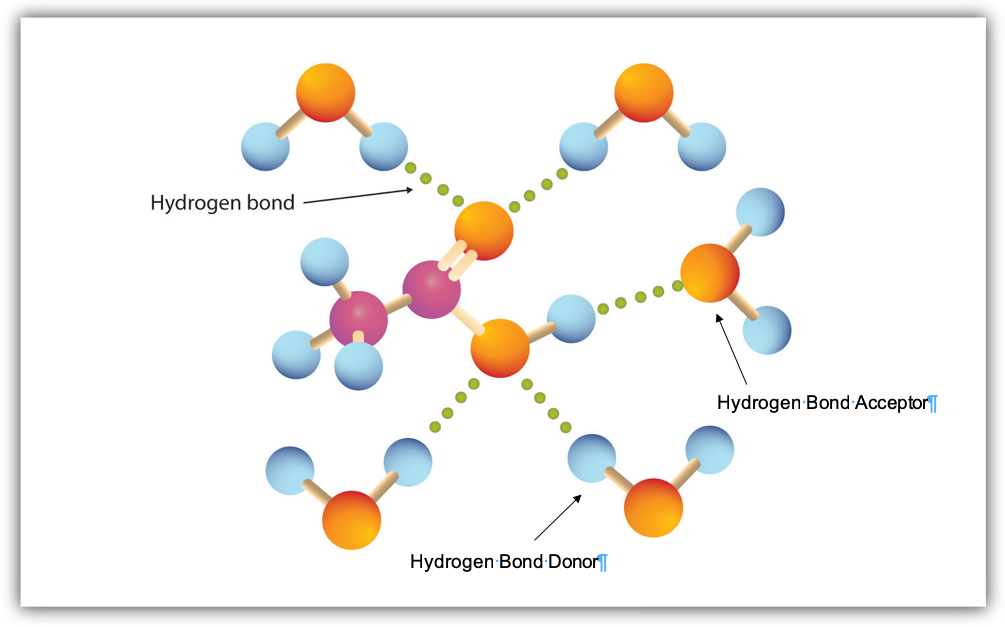
All these properties boil down to differing predictors of water vs. fat/lipid solubility. Generally speaking, if a molecule is too large it will likely be too lipophilic and not interact with the membrane the same way, and if there are too many hydrogen-bond donors or acceptors the molecule will be too polar or hydrophilic. As more and more drugs are developed and tested, we stumble across other classes of molecules that can enter cells but don’t conform to Lipinski’s rules. These are broadly termed beyond rule of 5 (bRo5) molecules.
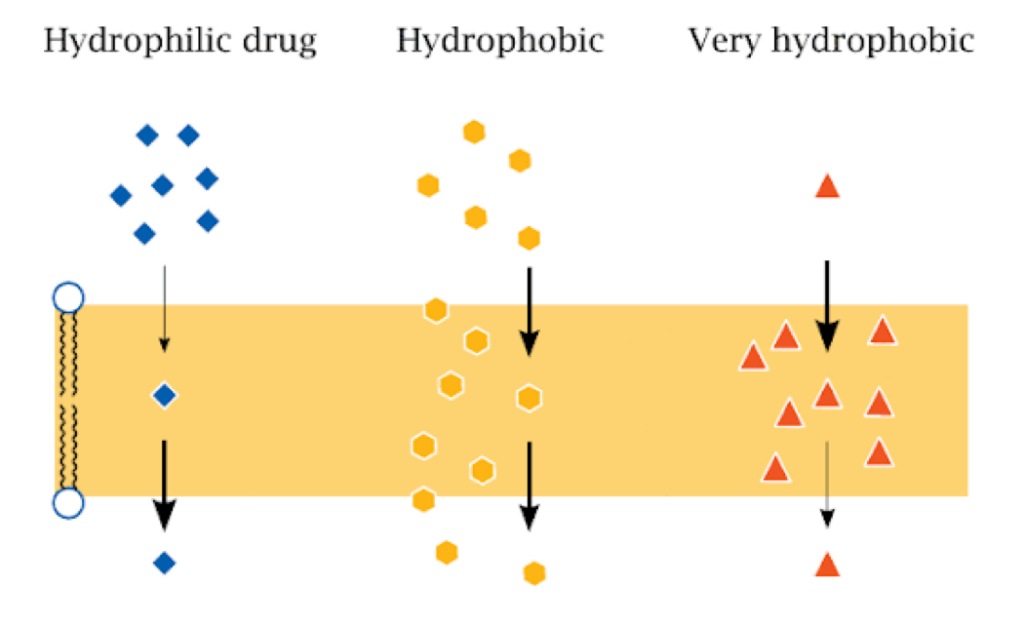
A general approach to make a drug more cell permeable is to remove or mask hydrogen-bond donors. Hydrogen bond donors in particular incur a high energy penalty when desolvated upon entering the lipid bilayer. The structure of a drug is generally highly optimised where each functional group serves a purpose, especially polar groups which often participate in key interactions with the target molecule. Consequently, masking or removing these groups to increase the cell permeability can decrease or eliminate the activity of the drug. A more favourable approach would be to mask the hydrogen bond donors with a group that can be removed once the drug has entered the cell. Even better, would be a mask that could be added to a completed drug molecule, so that the drug synthesis did not need to be commenced from the start or reoptimized. This was the goal of Barlow et al.
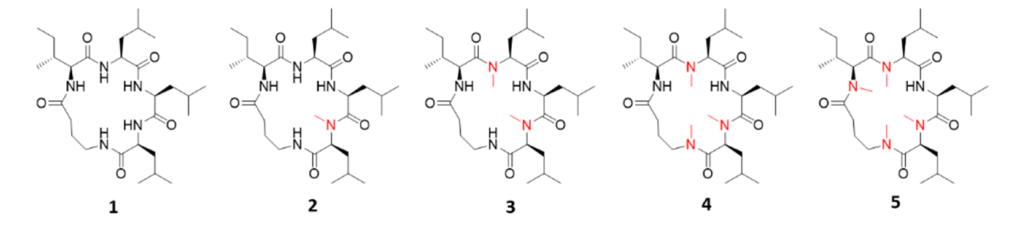
Peptides, strings of amino-acids, are particularly useful for targeting interactions between proteins as they are larger and consequently more specific than small molecules – this however limits their cell permeability and they are a classic example of a bRo5 molecule. Peptides are also full of different functional groups with HBAs and HBDs which makes them an ideal scaffold to investigate masking of functional groups to increase cell permeability. In this paper, a tetra-peptide cyclised with a short linker (1, figure below) was chosen. A common approach to reduce the number of HBDs in the HBD-rich amide backbone of a peptide is through N-methylation. Eight peptides with different N-methylation patterns were synthesised and their cell permeability tested by a PAMPA (Parallel Artificial Membrane Permeability Assay). This assay tests the ability of a molecule in solution to be able to passively cross an artificial membrane to a separate area and is measured in cm/s where a higher number indicates a higher permeability. The parent molecule 1 was almost impermeable, and the N-methylation screen demonstrated that at least two methylations were required to significantly improve the permeability. The position of the methylations didn’t have a large impact, likely owing to the flexibility of the peptide. The most permeable peptide 3, with two N-methylations, was chosen for further study.
The next stage was to mutate one of the leucine residues (a hydrophobic amino-acid) with a suite of other hydrogen-bond donor containing amino-acids to allow investigation of the masking effect. These eight peptides, with an HBD containing side-chain, were all essentially impermeable to the membrane which reiterates the large impact these groups play in membrane permeability. Next, to mask. The masking group formed an ester or carbamate group, or an acetamide in the case of lysine, to remove the HBD. These functional groups are not themselves important but what is important is that it instead forms a HBA which helped maintain the polarity required for aqueous solubility. These groups were specifically chosen as they’re present in a number of already FDA approved drugs. Furthermore, the linking group is cleavable under biological conditions, specifically by esterases (enzymes that break-down these types of groups) which are found in high abundance inside cells, which would allow the active molecule to be liberated upon entry. All of the masking groups were installed onto the already prepared parent molecule using a variety of common methodologies. All of the masked compounds were more permeable than parent molecule – reducing the HBDs did indeed improve the permeability of the peptides! Two masked peptides even had a greater permeability than the original leucine-containing peptide 3.
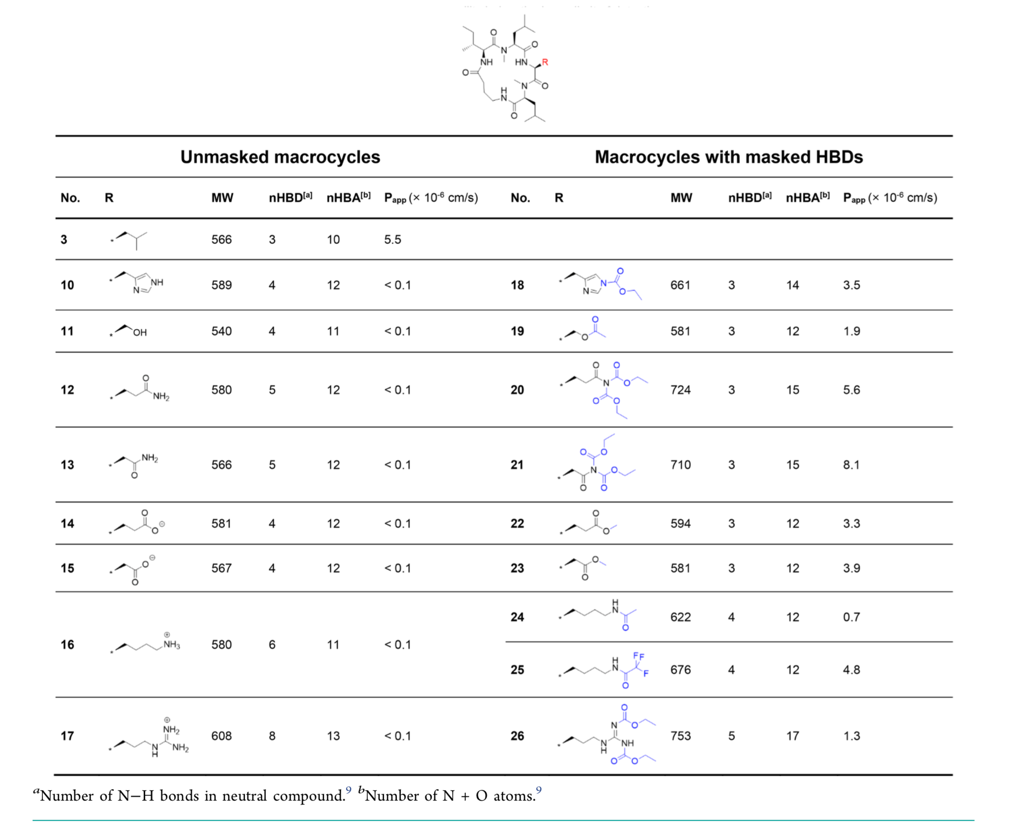
Overall, the masked peptides were stable, except for one which saw some liberation of the masking group over time. The authors noted these modifications were well tolerated and only saw solubility issues in peptides where the masking group introduced a lipophilic group such as in 13 to 22, and 14 to 23. The authors also confirmed that the permeability results from the artificial membrane in the PAMPA assay was comparable to a live-cell membrane in a Caco-2 test which tests the permeability of the molecules through a human colon epithelial cancer cell line.
Computational conformation searching of these molecules in both a water and chloroform (hydrophobic) environment demonstrated that the polar side-chains were well solvated in an aqueous environment and folded back across from macrocyclic peptide ring in chloroform so as to minimise exposure of the polar groups to the hydrophobic environment. This may be representative of the conformational changes that the peptide undergoes on transitioning from the aqueous phase through the membrane and back, and suggests that the increase in permeability of these molecules may be influenced by the conformations of the peptides and not only the reduced number of HBDs.
This study is a great example of how we can access bRo5 molecules and impart cell permeable character to drugs. This is an important technique that complements existing technology, and can be employed to pre-optimised structures to allows us to access functional and diverse scaffolds to drug new targets. For example, developing selective cancer therapeutics that have less side-effects than traditional chemotherapy such as peptide-based drugs that target the large surface area protein-protein interactions that dictate many biological processes.