

Article Title: Engineering of a Small Protein Scaffold to Recognize Sulfotyrosine with High Specificity
Authors: Lawrie, J.; Waldrop, S.; Morozov, A.; Niu, W.; Guo, J.
Journal: ACS Chem. Biol.
Year: 2021
DOI: /10.1021/acschembio.1c00382
After a protein is synthesized by the ribosome, it can often undergo further transformations known broadly as post-translational modifications (PTMs). These modifications include altering the side chains of specific residues, the N-termini, or C-termini by attaching small functional groups such as phosphates or even large biomolecules such as lipids and carbohydrates. Installation of these PTMs can greatly alter the activity of these proteins, and many cellular processes rely on these processes taking place. For instance, phosphorylation signal cascades help transmit and amplify signals to grow or respond to environmental stimuli. In other cases, protein cleavage can activate or inactivate enzyme activity. A final example is the attachment of a small protein, ubiquitin, to trigger protein degradation.
One particular PTM is protein tyrosine O-sulfation (PTS), or the appendage of sulfate from the universal donor PAPS to the oxygen atom of tyrosine (Figure 1). This occurs exclusively on transmembrane and secreted proteins, or proteins that wind up within the cell membrane or outside of the cell. It is believed that up to 1% of all tyrosine residues are sulfated.
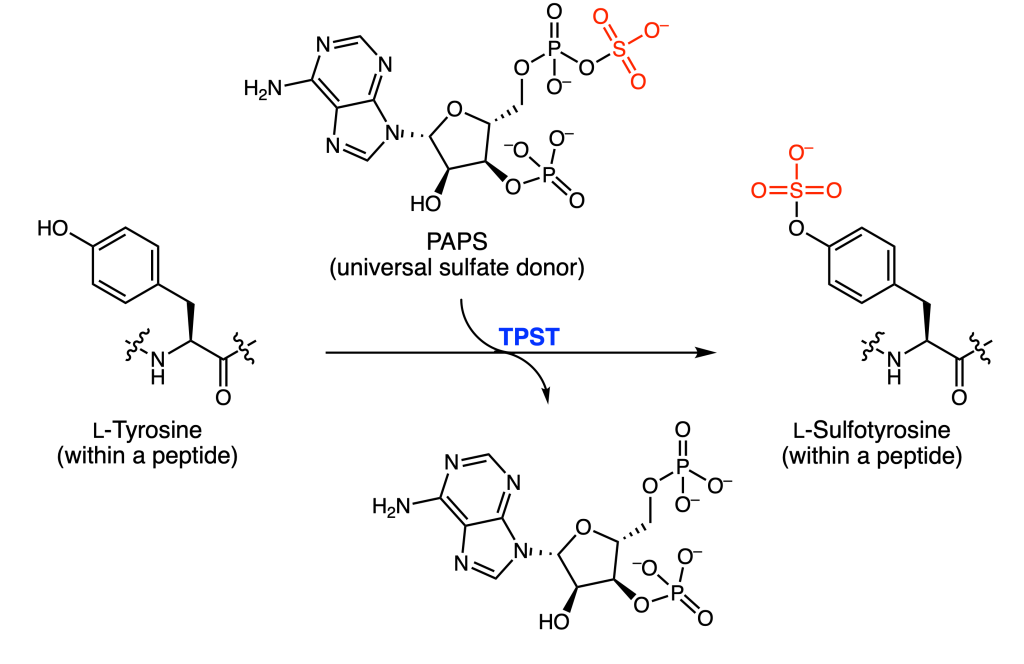
However, detection, enrichment, and characterization of these sulfotyrosines (sTyr) has been difficult. Although anti-sulfotyrosine antibodies are commercially available, their affinity for sTyr over a similar PTM, phosphotyrosine (pTyr), is small. To address this limitation, Lawrie and coworkers decided to engineer the protein domain Src Homology 2 (SH2), which normally recognizes and binds pTyr, to selectively bind sTyr instead (Figure 2). They targeted four residues (Arg35, Ser37, Glu38, and Thr39) that interacted with the phosphate group of pTyr and mutated them to all 20 possible amino acids.
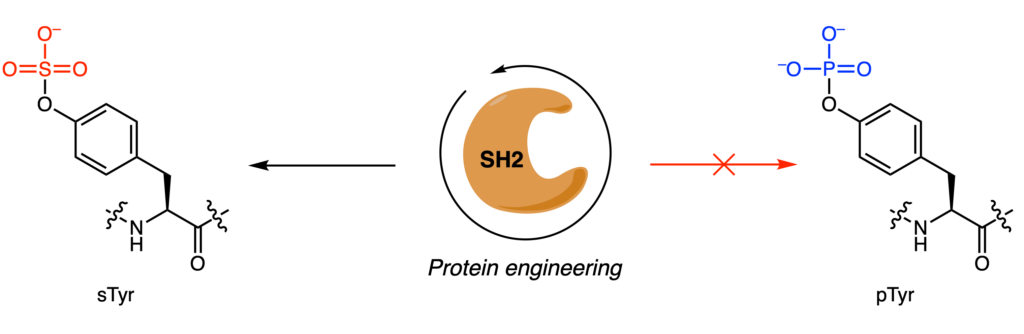
To screen this large library (1.05 • 106 mutants), the authors used phage display, a high-throughput screening technique that allows protein-protein interactions to be studied. Briefly, encoded proteins of interest can be expressed and “displayed” on the outside of bacteriophages. An analogy for this would be briefcases containing sheets of papers with instructions to decorate the exterior in a specific way. In this case, they hoped to detect interactions between a mutant SH2-domain and sTyr-peptides. Traditional phage display methods yielded no results, so they instead used a hyperphage system which enables five-copies of the protein to be expressed in 100% of the bacteriophages, rather than one-copy in 1% of bacteriophages. Three selection schemes were tested that prioritized either positive selection (binds sTyr), negative selection (does not bind pTyr), or competitive selection (binds sTyr over pTyr).
The binding affinity for the top ten mutants from each screen were measured. The only mutant with a binding affinity for sTyr over pTyr was from the third screening scheme. The authors speculate that negative selection (scheme two) was less successful because it removed variants with high affinity for sTyr and low affinity for pTyr. In general, mutants with improved sTyr affinity had an alanine rather than valine residue at position 37 and more basic residues. Comparative modeling of the mutants showed that better performing mutants had a reduced binding pocket. Since the electron density of sTyr is smaller than that of pTyr (sulfur is more electronegative than phosphorus), perhaps improved binding interactions enabled improved specificity.
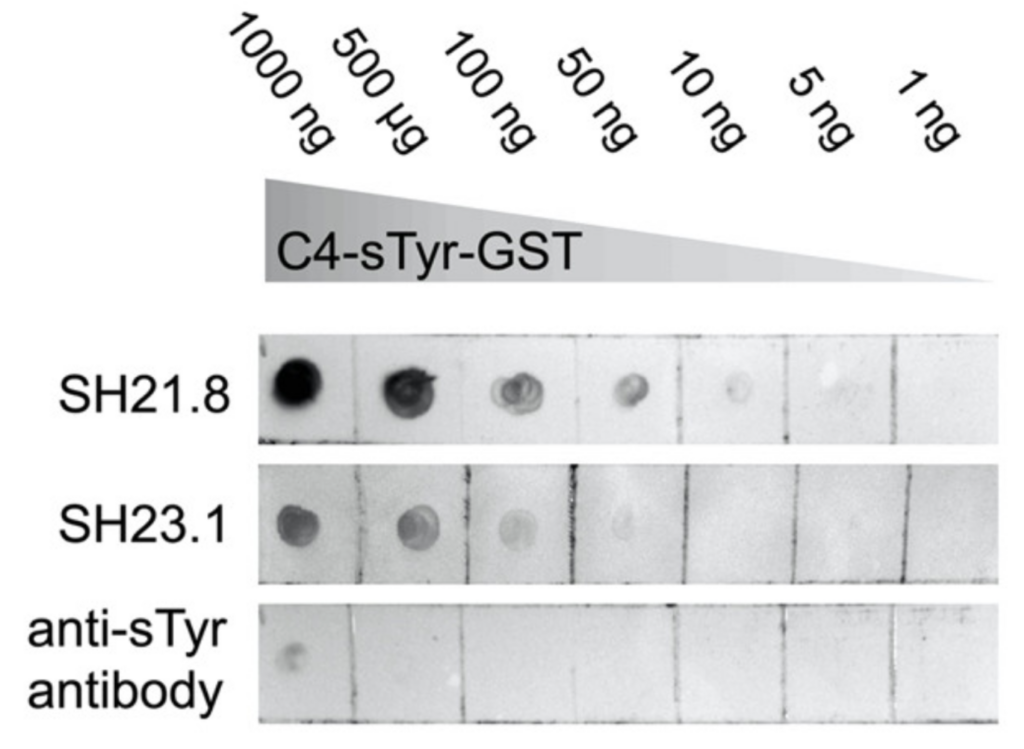
The utility of these newly acquired mutants was tested in dot blot analyses (Figure 3). Different amounts of a naturally sulfonated protein (C4-sTyr-GST) were tested against 2 mutants (SH2-1.8 and SH2-3.1) and compared to the commercial anti-sTyr antibodies. Based on the broad range of C4-sTyr-GST that was detected, SH2-1.8 had the highest affinity. In comparison, the commercial antibody was only able to detect sulfonated protein at the highest concentration. More complicated biological samples were also tested, and the mutants were once again able to enrich for known sulfonated proteins, even against a background of phosphorylated protein.
In this work, a promising alternative for selectively detecting sulfonated tyrosine over phosphorylated tyrosine was developed using a combination of protein engineering and phage display. In vitro experiments demonstrated that the sensitivity of these mutants was markedly improved over those currently offered commercially. Although these mutants can detect sulfonated proteins from complex samples, there is still some residual cross-reactivity with phosphorylated protein that limit their use for targeted enrichment and identification of novel sulfonated proteins. Continual work addressing these limitations could turn these SH2-mutants into valuable tools for chemical biologists.