

Featured image: Used with permission from Pixabay
| Title: | Crystal Structure Prediction via Deep Learning |
| Authors: | Kevin Ryan, Jeff Lengyel, and Michael Shatruk |
| Publication Info: | Journal of the American Chemical Society, 2018, Article ASAP DOI: 10.1021/jacs.8b03913 |
Machine learning? Deep neural networks? Whatever you’re thinking when you hear those words, I bet it isn’t chemistry! But the analytical power offered by modern-day technology is an enormous boon to chemists – including those who study crystallography, or the science of how atoms are arranged in a crystalline solid. Different crystals, like salt (NaCl) or diamond (C), arrange in different atomic patterns, which affect their chemical and physical properties. Crystallography helps define these differences and can help predict the chemical structure of new materials, from perovskites to zeolites.
Modern crystallography typically unites quantum mechanics (to evaluate the probability that a certain type of structure will be favorably formed) and some type of random initialization (that generates random values for atom position, to be evaluated by quantum mechanics). An efficient method of predicting crystal structures could help scientists discover new materials, but most current methods are costly in terms of time and computing power.
Here’s where deep neural networks come in. Deep neural networks are sets of algorithms, loosely based on the structure of biological brains, that help a computer recognize and interpret patterns. Deep neural networks help a computer “learn” to perform tasks without being specifically programmed to do so. They can simplify the task of figuring out how to turn crystallographic data into something that a computer can understand, saving researchers time and effort.
The paper discussed here demonstrates these advantages, taking atomic fingerprints (which describe the distance between nearest neighbors in a crystal structure) and using them to train the deep neural network. These atomic fingerprints contain data on the distances between atoms in a particular crystal structure type, like rock salt (Figure 1).
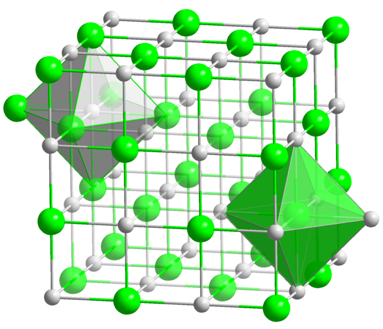
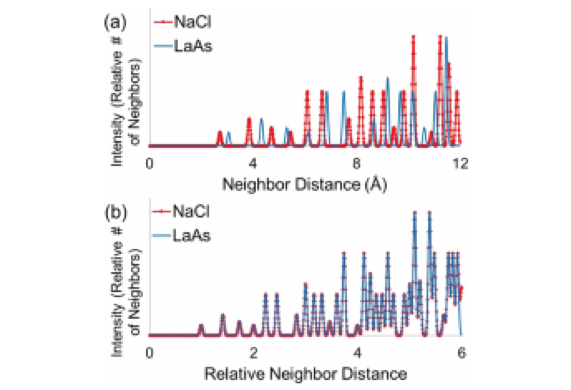
While atoms of different sizes have different absolute distances between them, relative distances stay the same for a given type of crystal (Figure 2). The deep neural network can therefore use data from a sample of a known structure to identify other materials with the same structure. This allows scientists to quickly evaluate a wide range of candidates for the crystal structure of a given material.
But what about predicting the crystal structure of a new material? Since most newly discovered crystal structures fall into categories that are already known, the authors of this paper use these known structure types as a starting point instead of random number generation. While they admit that this prevents the deep neural network from discovering new structure types, this part of the model can easily be replaced with a better method without interfering with the deep neural network’s ability to efficiently evaluate new structures.
New technologies like machine learning continually help advance science – including chemistry. The greatest challenge for researchers is figuring out how to adapt these tools to their own fields, but once adapted, they can yield impressive results.